


Высококачественные наноалмазы с зеленым излучением, полученные спеканием поликристаллических ударно-волновых алмазов методом HPHT
Аннотация
Мы демонстрируем технику спекания при высоком давлении и высокой температуре для образования центров азот-вакансия-азот в наноалмазах. Поликристаллические предшественники алмазных наночастиц со средним размером 25 нм образуются ударной волной от взрыва. Эти наночастицы спекаются в присутствии этанола при давлении 7 ГПа и температуре 1300 ° C с получением значительно более крупных (в 3–4 раза) кристаллитов алмаза. Зарегистрированные спектральные свойства демонстрируют улучшенное кристаллическое качество. Наблюдается также изменение типов имеющихся дефектов; характерные спектральные особенности центров вакансий азота и кремния, присутствующие в материале-прекурсоре, исчезают. Появляются две новые характерные особенности:(1) парамагнитный азот замещения (центры P1 со спином 1/2) с характерной для электронного парамагнитного резонанса триплетной сверхтонкой структурой, обусловленной I =1 магнитный момент ядерного спина азота и (2) зеленая спектральная характеристика фотолюминесценции центров азот-вакансия-азот. Этот метод производства является сильной альтернативой традиционному облучению пучком частиц высокой энергии. Его можно использовать для простого производства чисто зеленых флуоресцентных наноалмазов с полезными свойствами для применения в оптических биомаркировках.
Введение
Примеси и вакансии азота являются преобладающими дефектами в большинстве природных и синтетических алмазов. Индивидуальные дефекты могут коллективно образовывать комплексы дефектов, содержащие до 6 субъединиц [1, 2]. Из этих дефектных комплексов азот-вакансия (NV - ) и, в меньшей степени, центры азот-вакансия-азот (NVN) привлекли значительный интерес из-за их немигающей красной и зеленой фотолюминесценции соответственно [3, 4]. NV - и NVN можно контролируемо генерировать в наноалмазах. Наноалмазы широко признаны нетоксичными наночастицами и, следовательно, могут использоваться в качестве долгосрочных отслеживаемых меток в биомедицинских приложениях [5]. Нанокристаллы с НВ - центры окраски также используются для квантового зондирования [3].
Синтез алмазов при высоком давлении и высокой температуре (HPHT) с использованием традиционных катализаторов на основе растворителей переходных металлов является стандартной промышленной технологией. Он используется во многих лабораториях для выращивания кристаллов алмаза с улучшенными параметрами решетки. Однако в последние годы стали широко применяться различные нетрадиционные металлические катализаторы [6]. Такой метод позволяет использовать азотсодержащие органические добавки и газопоглотители для контролируемого легирования алмаза в диапазоне от высокого содержания азота (до ~ 1000 ppm) до гораздо более низкого уровня (~ 50 ppm) [7, 8] . HPHT также используется для отжига кристаллов алмаза и улучшения их кристаллического качества, для обесцвечивания и спекания нанокристаллов в более крупные поликристаллы.
Группирование индивидуальных дефектов азота в более крупные комплексы в алмазе под влиянием температуры (и давления) широко изучалось. Такие комплексы могут служить специфическими маркерами, фиксирующими температурную историю завершенных процессов кристаллизации [1]. NVN и NV - центры могут быть созданы путем облучения чистых азотсодержащих алмазов высокоэнергетическими (2–14 мэВ) электронами или протонами или тяжелыми ионами с энергией ~ 40 кэВ. Облучение создает вакансии в решетке алмаза. Последующий отжиг образцов при температурах в диапазоне 500–2000 ° C вызывает группировку дефектов [3, 9, 10]. В настоящее время серийное производство НВ - или наноалмазы, содержащие NVN, обычно получают путем облучения электронами высокой энергии. Однако этот процесс дорог и трудоемок, а также требует специальной пробоподготовки. Таким образом, стоит разработать альтернативные методы создания центров окраски в наноалмазах, не требующие облучения частицами.
Недавно был предложен новый метод изготовления кристаллов алмаза субмикронных размеров, основанный на спекании 5-нм детонационных наноалмазов (ДНА) в присутствии жидких добавок C – H – O [11, 12]. В условиях спекания HPHT добавленные органические соединения C – H – O ведут себя как сверхкритическая жидкость, которая вызывает быструю рекристаллизацию алмаза. Быстрая рекристаллизация сопровождается увеличением плотности вакансий [13]. Однако этот метод подавляет первичные центры окраски, присутствующие в предшественниках 5-нм ДНА [14]. Следовательно, желателен альтернативный подход, который может сохранить эти дефекты или даже создать другие типы в процессе спекания HPHT.
В данной работе мы используем поликристаллические наноалмазы-прекурсоры, полученные взрывным методом с использованием так называемой технологии ударно-волнового синтеза DuPont [15,16,17]. Мы спекаем эти наноалмазы в присутствии добавок C – H – O так же, как в существующей запатентованной методике [18] (рис. 1), получая крупные субмикронные алмазы, содержащие только центры окраски NVN, которые не присутствовали в прекурсоре. наночастицы. Мы также отслеживаем эволюцию всего NV - , P1 и SiV-центры в частицах до и после спекания с использованием электронного парамагнитного резонанса (ЭПР) и флуоресценции, чтобы продемонстрировать влияние спекания на весь спектр имеющихся дефектов.
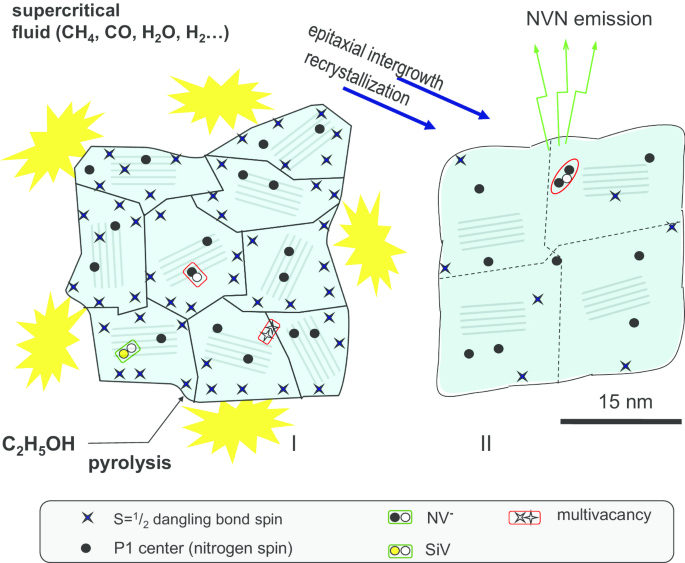
Схематическое изображение спекания поликристаллических алмазных частиц в условиях высокого давления и высоких температур с получением субмикронных нанокристаллов алмаза с размерами около 30 нм. Различные дефекты и центры окраски наблюдаются как в поликристаллах (I), так и в нанокристаллах после спекания HPHT (II)
Методы
Образец изготовления
Субмикронные кристаллы алмаза получали спеканием коммерчески доступных исходных поликристаллических наноалмазов со средним размером 25 нм (продукт DP 0–0,05, Microdiamant, Швейцария). Частицы прекурсора получают с помощью взрывной техники, при которой происходит быстрое преобразование графита в алмаз, вызванное ударной волной (в течение ста микросекунд при T ≈ 950 ° С, P ≈ 50 ГПа). Продукты, образовавшиеся в результате взрыва, впоследствии подвергаются химической обработке для очистки фазы наночастиц алмаза, удаления остаточных металлов и аморфного углерода. Порошок состоял из крупных поликристаллов размером> 10 мкм. Эти поликристаллы можно легко фракционировать путем измельчения и дезагрегации по границам зерен, следуя ранее описанному методу [19]. Для выделения фракции малого размера, образующей поликристаллы DP 0–0,05, было проведено фракционирование по размеру. Полученное распределение по размерам (рис. 2) для самой мелкой фракции имеет максимум при 27 нм и полуширину на полувысоте (FWHM) ≈ 25 нм (95% частиц имеют размеры в диапазоне 3–50 нм). Затем выбранные частицы прекурсора переносятся во внутренний графитовый цилиндр камеры высокого давления тороидального типа для спекания. Типичные размеры внутренней части камеры высокого давления (т.е. вышеупомянутого полого графитового цилиндра с двумя графитовыми крышками) составляли:внутренний диаметр 4,0 мм и высота 5,5 мм. Перед спеканием к сухому порошку наноалмаза по каплям добавляли этанол до полного заполнения межчастичного пространства (около 30–50 мас.%) [18]. Затем спекание происходило в условиях высокого давления (7 ГПа) и высоких температур (1300 ± 50 ° C) в течение 10 с. За одно нажатие можно было обработать около 120 мг исходного алмазного порошка. Схема камеры высокого давления приведена в [5]. [11].
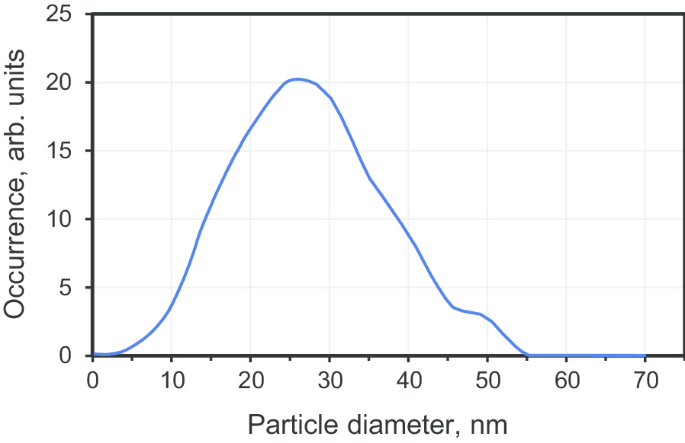
Распределение по размерам фракции поликристаллического алмаза DP 0–0,05 со средним размером ~ 27 нм, измеренное с помощью аппарата дифференциального центробежного осаждения (дисковая центрифуга CPS, CPS Instruments Inc., США)
Эффект этого процесса спекания показан на рис. 1. При спекании в условиях HPHT этанол находится в сверхкритическом состоянии. Следовательно, он может легко проникать через границы поликристаллических зерен, способствуя рекристаллизации алмаза и росту новой алмазной фазы. После спекания ~ 90–100 мг белого алмазного порошка приемлемого качества (без загрязнения графитовым контейнером) было извлечено из камеры высокого давления (обозначенной здесь D19). Изменение цвета порошка с серого / черного прекурсора на белый после спекания указывает на изменение поверхности поликристаллов и их соответствующий рост [11].
Структурные характеристики материалов до и после спекания
Структурная характеристика предшественника поликристаллического наноалмаза
Мы сначала охарактеризовали структуру DP 0–0,05 перед спеканием. Измерения дифракции рентгеновских лучей (XRD) проводили с помощью рентгеновского дифрактометра Rigaku Smart Lab III с использованием излучения CuKα ( λ =1,54178 Å). Использовались напряжение 40 кВ, ток 30 мА и скорость сканирования 0,1 градус / мин. Картина XRD DP 0–0,05 показана на рис. 3 (черный) для 2 θ в диапазоне от 6 ° до 155 °. Мы наблюдаем шесть пиков, пять из которых (111, 220, 311, 400 и 331) характерны для алмазной фазы углерода. Самый сильный наблюдаемый пик - это кубический алмаз (D c ) 111 при 2 θ ≈ 43,7 °. Эта вершина не симметрична и представляет собой левое плечо. Подгонка образца к двум лоренцевам дает первый пик с центром в 2 θ =43,76 ° и секунда при 2 θ =41,64 °. Первый пик соответствует отражению от алмазных плоскостей (111), а второй (обозначенный D H 011) предположительно приписывают либо:(а) отражениям от плоскостей гексагональной 6H-политипной алмазной фазы [20], либо (б) множественным дефектам упаковки, двойникам и связанным границам зерен между кристаллитами под высоким напряжением. Сравнивая интегралы от D H 011 и D c 111, по нашим оценкам, неидентифицированная «фаза» (гексагональная или состоящая из структурных дефектов) в прекурсоре DP 0–0.05 составляет ≈ 20 мас.%. Пик, обозначенный 002 Graphite (при 2 θ ≈ 26 °) относится к фазе нанографита. Хотя эта примесная фаза может быть легко удалена, мы намеренно не травили кислотой в этом исследовании. Путем сравнения площадей пиков 002 и 111 было установлено, что доля этой аморфной фазы составляет ~ 4 мас.%. Затем мы оценили длину области когерентного рассеяния ( L CSR ) с помощью метода Шерера. Мы рассмотрели FWHM β пяти алмазных пиков в зависимости от sec θ . Β определяется после деконволюции из функции отклика дифрактометра. Footnote 1 L CSR для образца DP 0–0,05 оказалось равным 6,7 нм.
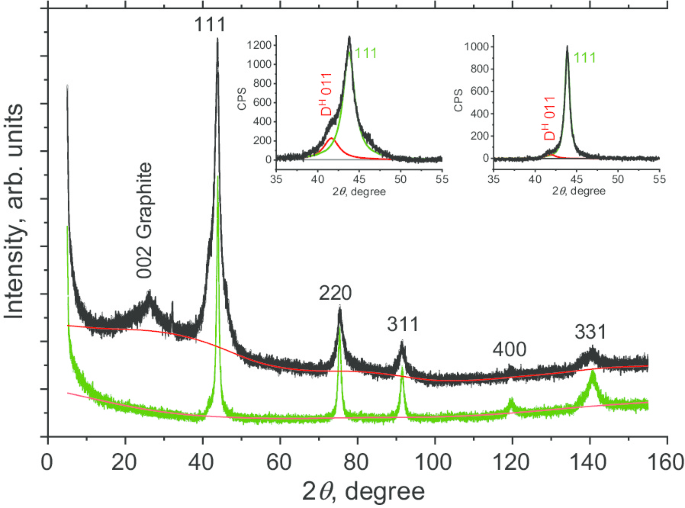
Профили порошковой XRD фракции DP 0–0,05 поликристаллических алмазных частиц (черная кривая) и наноалмазов D19, полученных спеканием (зеленая кривая). Вставки:Подгонка наиболее интенсивного отражения от плоскостей (111) двумя (красной и зеленой) кривыми Лоренца с центром под углом 41,5 ° (D H 011) и 43,8 o (D c 111) для образцов до (левая вставка) и после спекания (правая вставка)
Чтобы получить более полное представление о сложной структуре поликристаллов алмаза DP 0–0,05, мы использовали просвечивающий электронный микроскоп высокого разрешения (HRTEM, JEOL JEM-2100F, оснащенный корректорами CEOS Cs, Университет Шиншу). HRTEM работал при 80 кВ, чтобы минимизировать радиационные повреждения. Изображения ПЭМВР (рис. 4) показывают, что поликристаллы состоят из прочно связанных кубических кристаллитов наноалмаза с характеристикой 1,93 Å d -бахрома. Поликристалл, изображенный на рис. 4а, состоит из кристаллитов размером от 5 до 12 нм, имеющих разную ориентацию, отмеченных разными цветами. Кроме того, ПЭМВР демонстрирует, что размер отдельных поликристаллов, составляющих порошок DP 0–0,05, широко распределяется в диапазоне 5–50 нм. Изображения HRTEM не выявили никаких следов гексагональной нанофазы алмаза (например, лонсдейлита). Однако мы наблюдаем множество коротких границ двойникования (рис. 4c, d), некоторые из них демонстрируют гофрированные плоскости решетки с формой гармошки, как на рис. 4d. Эти наблюдения показывают, что плечо пика 111 XRD (рис. 3), обсуждавшееся ранее, скорее всего, связано с дефектами упаковки, связанными с множественными границами зерен, связанными с двойникованием, а не с наличием гексагональной фазы алмаза [16].
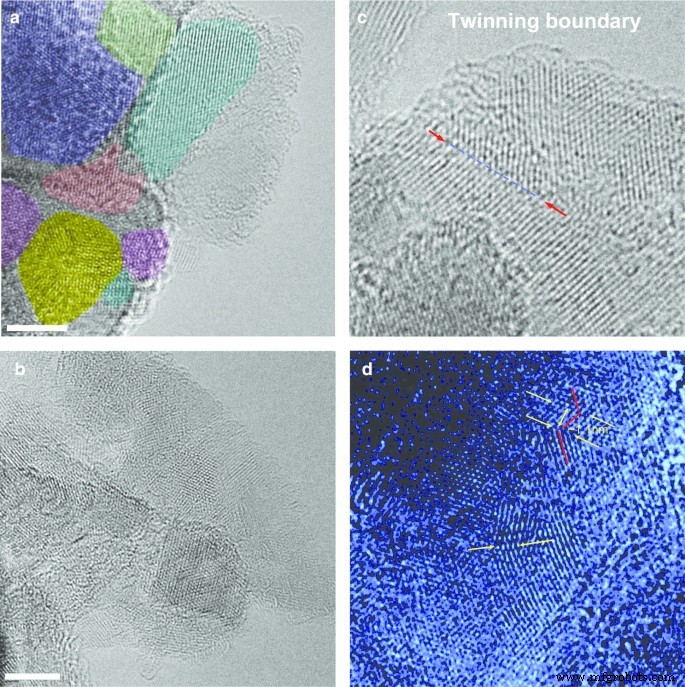
ПЭМ-изображения с высоким разрешением выбранных кубических поликристаллов алмаза, извлеченных из порошка DP 0–0,05 ( a , b ) и типичные изображения простых границ двойникования длиной несколько нанометров, которые иногда встречаются в образце ( c , d ). Панели ( a , b ):масштабная линейка - 4 нм. Различные кристаллиты выделены разными цветами на ( a ). Стрелки на панелях ( c ) и ( d ) отметить выбранные четко различимые границы двойникования
Элементный состав DP 0–0,05 анализировали методом Прегля – Дюма с использованием системы элементного органического анализа (JM10, J-Science Lab Co., Ltd, Киото, Япония), в которой образец обжигали при 1007 ° C в поток кислорода (30 мл / мин). Долю кислорода определяли на весах. Результаты анализа в мас.%:C - 90,45, N - 2,47, H - 0,76, O - 6,32. Относительно высокая концентрация азота означает, что почти весь азот присутствует внутри поликристаллов в агрегированной форме, вероятно, в форме A-центров (NN-димеров). Дальнейший анализ с помощью рентгеновской флуоресценции показал присутствие других микроэлементов в DP 0–0,05:Fe (~ 300 частей на миллион), Cu (35 частей на миллион), Si (120 частей на миллион), Cr (~ 150 частей на миллион), Ca (~ 45 частей на миллион). ppm), Mn (45 ppm), P (30 ppm), Al (18 ppm), Ti (13 ppm), Mg (6 ppm), Ni (2 ppm), Zn (2 ppm), Co (1 ppm) .
Образец DP 0–0,05, использованный для последующих исследований магнитного резонанса, был дополнительно обработан кипящей соляной кислотой для снижения содержания ферромагнитных металлов (в основном железа и хрома) до уровня ~ 10 ppm.
Структурная характеристика спеченного алмаза
Аналогичным образом был охарактеризован образец Д19, полученный спеканием DP 0–0,05. Рентгенограмма D19 была получена тем же методом, что и для порошка DP 0–0,05. Было обнаружено, что отображены только пять пиков лоренцевой формы, характерных для кубической алмазной фазы (рис. 3, зеленая линия), соответствующих плоскостям 111, 220, 311, 400 и 331. Нет никаких доказательств присутствия графитовой фазы в исходном материале. Диссимметрия пика 111 также сильно уменьшена (рис. 3, правая вставка). Это говорит о соответствующем значительном сокращении дефектов упаковки. Кроме того, L CSR Длина когерентности составила 10,8 нм, что указывает на увеличение нанокристаллитов алмаза во время спекания. Принимая во внимание эти наблюдения, мы предполагаем, что графитная и некубическая алмазные фазы превращаются в кубическую алмазную фазу в процессе спекания. Это, вероятно, является результатом растворения этих фаз с последующим ростом кубической алмазной фазы в межузельных пространствах между кристаллитами поликристаллов. Такой процесс ранее наблюдался для детонационных наноалмазов [14].
Изображения образца D19, полученные с помощью оптической и растровой электронной микроскопии, представлены на рис. 5а, б соответственно. Здесь мы видим частицы произвольной формы размером от ~ 0,3 до ~ 5 мкм. На самом деле они представляют собой плотные субмикронные и очень рыхлые агрегаты микронных размеров гораздо более мелких алмазных кристаллитов, связанных друг с другом ковалентными связями или слабыми ван-дер-ваальсовыми связями. Такие связи обычно возникают в результате взаимодействия поверхностных функциональных групп соседних алмазных частиц. Отдельные алмазные частицы, составляющие эти рыхлые агрегаты, можно увидеть с помощью просвечивающей электронной микроскопии. ПЭМ-изображение кристаллита алмаза D19 размером около ~ 100 нм показано на рис. 5в. Это изображение было получено с помощью просвечивающего электронного микроскопа JEOL JEM-2100F (Университет Хосей) при ускоряющем напряжении 200 кВ. Образец закреплялся на медной сетке без угольной подложки.

Оптический ( a ), сканирующая электронная микроскопия ( б ) и просвечивающая электронная микроскопия ( c ) изображения синтезированных алмазных частиц D19. Оптическое изображение было получено с помощью 100-кратного микроскопического объектива. Соответствующие параметры, используемые для получения электронного SEM-изображения, указаны в нижней части панели ( b )
Методы анализа содержания дефектов в предварительно и пост-спеченных наноалмазах
Оба образца алмаза были проанализированы с помощью спектроскопии ЭПР, комбинационного рассеяния света и флуоресценции. Спектры ЭПР регистрировали при комнатной температуре на частоте микроволн 9,444 ГГц с использованием спектрометра ЭПР (JES-FA 300, JEOL, Япония). В кварцевую трубку ЭПР диаметром 4 мм вводили 20 мг порошка. Высота столба порошка в трубке не превышала 10 мм. Открытый конец трубки был защищен от влаги.
Спектры ЭПР с g -факторы в диапазоне g =4.00–4.30 были зарегистрированы при мощности микроволн P МВт =10 мВт, амплитуда модуляции магнитного поля A м =1 мТл и частота ν =100 кГц, усиление G ≈ 10 3 , и N =16 циклов накопления сигнала. Эти параметры были выбраны для получения оптимального отношения сигнал / шум. Постоянная времени составляла 0,03 с, а общее время регистрации развертки магнитного поля в интервале 130–200 мТл составляло 120 с. Спектры ЭПР с g -factors g ≈ 2 были зарегистрированы в интервале от 327 до 347 мТл при мощности микроволн P МВт =0,03 мВт, амплитуда модуляции магнитного поля A м =0,035 мТл, усиление усилителя G ≈ 10 2 , и N =4 цикла накопления сигнала. Обратите внимание, что, как правило, для широких основных сигналов ЭПР ( g ≈ 2) с шириной линии> 0,5 мТл, размах сигнала ЭПР ( I pp ) следует зависимости мощности от МВ I pp ~ ( P МВт ) 1/2 до P МВт ≈ 100 мВт. С другой стороны, для узких сигналов ЭПР (ширина линии <0,15 мТл при малой мощности) I pp насыщается при P МВт > 0,05 мВт и имеет сильное искажение формы при более высоких значениях (> 4 мВт). Такие тенденции насыщения наблюдались как для поликристаллического прекурсора, так и для впоследствии спеченных алмазов.
Мы получали спектры фотолюминесценции (PL) и комбинационного рассеяния света с помощью спектрометра микро-комбинационного рассеяния («inVia», Renishaw, Великобритания) в сочетании с оптическим микроскопом (Leica, Германия) с использованием объектива 50x (NA =0,78) и ПЗС-матрицы. детектор, охлаждаемый до -70 ° C, с геометрией обратного рассеяния. Спектры записаны со спектральным разрешением ~ 2 см –1 . . Мы использовали две лазерные линии аргон-ионного лазера на длинах волн 488 нм и 457 нм с интенсивностью менее 20 Вт / см −2 . в фокусе на образце. Мы записали спектральные изображения в режиме StreamLine ™ Plus (Renishaw, UK), который использует более низкую интенсивность возбуждающего лазера в плоскости образца из-за его фокусировки на полосе 2 × 30 мкм по сравнению со стандартной фокусировкой на круге микронного размера. Эта стратегия ограничивала лазерное повреждение образца и локальное газофазное травление из-за перегрева и окисления в окружающей атмосфере. Дополнительные подробности этого метода и прессования алмазного порошка в цилиндрах диаметром 2 мм были описаны ранее [19].
Флуоресцентные изображения изолированных частиц D19 получали с помощью конфокальной широкопольной эпифлуоресцентной микроскопии с объективом 100 ×. Частицы осаждали на покровное стекло из фракции супернатанта разбавленной водной суспензии порошка D19 посредством центрифугирования. Покровное стекло предварительно обрабатывали кислородной плазмой, чтобы избежать паразитической флуоресценции от остаточной органики и способствовать лучшему прикреплению частиц D19 к покровному стеклу. Флуоресценцию возбуждали лазером с длиной волны 488 нм (мощность возбуждения ≈ 40 мВт) и собирали с помощью интерференционного фильтра 525/40. Для записи изображений использовался специально охлаждаемый 2D-матричный ПЗС-детектор (температура камеры =-79,9 ° C). Изображения (размер ~ 80 × 80 мкм, 80,00 нм / пиксель) были записаны с выдержкой 60 мс. Для представления изображения использовалась палитра оттенков серого. Изображение было проанализировано с помощью программного обеспечения Fiji.
Результаты
ЭПР поликристаллов и кристаллитов алмаза до и после спекания
Спектры ЭПР поликристаллического алмазного порошка DP 0–0,05 как в диапазоне половинного магнитного поля, так и в диапазоне сильного поля показаны на рис. 6. Спектры ЭПР, зарегистрированные в этих различных диапазонах, были измерены при высоком и низком микроволновом диапазоне. мощности P МВт =10 мВт и P МВт =0,03 мВт соответственно. Здесь область сильного поля обычно выбиралась вблизи линии поглощения, относящейся к основному микроволновому триггеру Δ M s =1 переход спинов S =½, тогда как расширенный диапазон полуполя был специально выбран для поиска сигналов от Δ M s =2 перехода возможных триплетов S =1 центр. Полуполевой спектр ЭПР имеет очень низкую интенсивность и демонстрирует наличие триплета NV - центры (сигнал с g =4,27 при H res =158 мТл [10, 16]) и триплетной мультивакансии (сигнал с g =4,00 при H res =168 мТл [16]) в прекурсоре поликристаллического алмаза. Оба этих типа дефектов присутствуют в очень малых концентрациях (<1 ppm); следовательно, для обнаружения таких редких центров необходимо было использовать высокую микроволновую мощность. Более того, в области сильного поля мы также наблюдали характерную сигнатуру спин-полуцентров ( g =2,0027 при H res =337 мТл [10, 16, 21]), но с широким (Δ H pp =0,47 мТл), форма одной производной Лоренца без какой-либо тонкой структуры [16]. Принимая во внимание предположение, что эти спины происходят из независимых центров, этот широкий интенсивный сигнал можно условно приписать спинам оборванных связей C – C и обменным парам парамагнитных спинов азота с неразрешенной сверхтонкой структурой (HFS). Footnote 2 Мы оценили концентрацию всех парамагнитных частиц со спином S =1/2 будет ~ 4 × 10 19 спин / г (800 частей на миллион), что в ~ 1,5 раза меньше, чем ранее сообщалось для детонационных наноалмазов 5 нм [22].
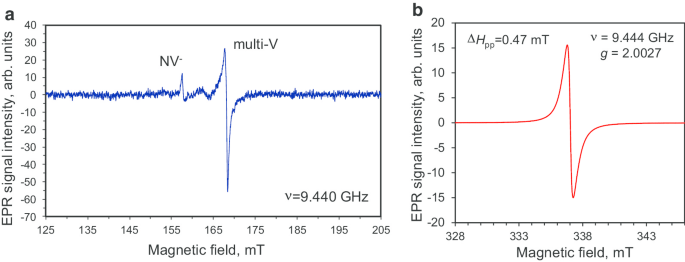
Спектры ЭПР фракции DP 0–0,05 алмазных частиц в диапазоне половин магнитного поля ( a ) и вокруг резонансного магнитного поля синглета с сильным сигналом при g -factor g ≈ 2,0027 ( b ). Мощность микроволн P МВт :10 мВт ( a ) и 0,03 мВт ( b ). Частота микроволн ν =9,44 ГГц. Оба спектра зарегистрированы в режиме, далеком от насыщения
На рис. 7а показаны спектры ЭПР образца D19 для очень низких ( P МВт =1 мкВт) и очень высокой (200 мВт) мощности микроволн, т.е. в режимах намного ниже насыщения или при насыщении (о чем свидетельствует уширение) соответственно. Спектр для P МВт =1 Wμ имеет центральную линию с более узкой шириной 0,14 мТл по сравнению с линией исходных частиц. На рис. 7б (кривая 1) показан дважды интегрированный сигнал ЭПР малой мощности СВЧ. Этот сигнал можно разложить на:широкий (FWHM ≈ 1,7 мТл) сигнал лоренцевой формы (рис. 7b, кривая 2), связанный с наиболее распространенным S =1/2 частиц (высокой локальной плотности и организованных в спиновые кластеры) и очень узкая центральная линия с двумя симметричными линиями-сателлитами, разделенными ~ 6 мТл (рис. 7б, кривая 3). Отношения интегральных интенсивностей для двух спутниковых линий относительно центральной линии равны 0,90 и 1,09 соответственно. Следовательно, они имеют равные интегральные интенсивности с точностью экспериментальных измерений ± 10%. Спутниковые линии связаны со сверхтонкой структурой сигнала ЭПР нейтрального парамагнитного замещающего азота ( 14 N, S =1/2, I =1), известные как центры P1 [23, 24]. Центры P1 не наблюдались в исходных поликристаллических наноалмазах, но четко обнаруживаются в алмазе после спекания. Предыдущая работа показала, что эта характерная триплетная структура СТС, связанная с центрами P1, проявляется только в крупных кристаллах алмаза размером более 50–80 нм [25]. Более того, было продемонстрировано, что спекание детонационных наноалмазов в присутствии этанола способствует увеличению кристаллитов на основе рекристаллизации алмаза и роста новой алмазной фазы [26]. Следовательно, текущие данные ЭПР согласуются с аналогичным увеличением, происходящим здесь от отдельных кристаллитов поликристаллических частиц размером ~ 7-10 нм до новых кристаллитов индивидуального размера ~ 40-50 нм. Footnote 3
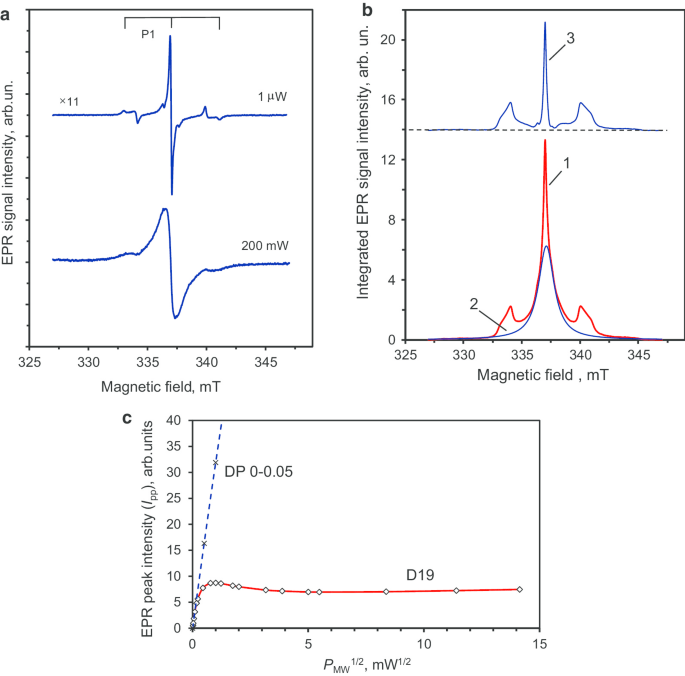
Основной сигнал ЭПР первой производной кристаллов алмаза D19 при низкой и высокой мощности СВЧ ( a ), разложение интегрированного сигнала ЭПР на компоненты, относящиеся к двум группам спинов ( b ), и соответствующая тенденция насыщения пиковой интенсивности этого сигнала ЭПР в зависимости от квадратного корня из микроволновой мощности в диапазоне до 200 мВт ( c ). Триплетная HFS-структура сигнала ЭПР центра P1 (замещающий азот) четко различима в ( a ). В ( b ):кривая 3, имеющая триплетную структуру, соответствует спектру ЭПР только центров P1. В ( c ):пунктирная прямая линия - это I pp vs ( P МВт ) 1/2 Для поликристаллов DP 0–0,05 зависимость приведена для справки. Четыре экспериментальные точки на P МВт =0.5, 1, 2, 4 мВт (два из них здесь не приводятся). Частота микроволн ν =9,44 ГГц
Эта гипотеза подтверждается не только наблюдением узкого сигнала ЭПР Footnote 4 ( г =2.0024) центров P1, обладающих характеристиками СТС [26], но также по обнаружению I pp насыщенность выше P МВт =0,7 мВт для образца D19. Для предшественника поликристаллического наноалмаза DP 0–0,05 пиковая интенсивность основного сигнала ЭПР не достигает насыщения даже при высоком уровне микроволновой мощности ( P МВт =20 мВт). Степенная зависимость I pp ~ ( P МВт ) 1/2 хорошо держится во всем диапазоне мощности СВЧ ( P МВт =0–20 мВт). Я pp ~ ( P МВт ) 1/2 Зависимость для образца DP 0–0.05 показана на рис. 7в штриховой линией до мощности СВЧ ~ 16 мВт. Такая линейная зависимость без насыщения является результатом высокой концентрации парамагнитных центров в поликристаллических наноалмазах и очень коротких времен спин-решеточной и спин-спиновой релаксации. Поведение насыщения образца D19 g =2.0024 Сигнал ЭПР также отображается на рис. 7c в диапазоне 0–200 мВт. Сноска 5 Мы наблюдаем Я pp ~ ( P МВт ) 1/2 зависимость только для СВЧ мощности ниже ~ 15 Втμ. В этом случае Я pp демонстрирует насыщение в диапазоне ниже 1 мВт и достигает максимума при ~ 1 мВт. Я pp затем существенно уменьшается в диапазоне 1–25 мВт перед тем, как снова медленно увеличиваться до максимальной используемой мощности (200 мВт). Наличие такого тренда насыщения в I pp ( P МВт ), а также капля над P МВт =1 мВт, характерно для центров P1 с относительно большими временами спин-спиновой и спин-решеточной релаксации, удаленных от других дефектов и от краев частиц [27]. Эти условия выполняются для концентраций центров P1 менее 200 ppm в наноалмазах HPHT типа Ib с размером более 60–80 нм. Однако фактический средний размер отдельных кристаллитов алмаза D19 (элементарных частиц), если смотреть только с точки зрения ЭПР, остается открытым вопросом. Ее можно приблизительно решить, сравнивая реальный спектр ЭПР D19 с серией порошковых спектров ЭПР измельченных HPHT-алмазов Ib со средним размером от 18 до 390 нм [28]. Следуя Ref. [28], где были опубликованы эти спектры ЭПР, HFS-сигнатуры P1, связанные с замещающим азотом, полностью отсутствуют в порошковых алмазах со средним размером ≤ 30 нм, но все же присутствуют в образцах промежуточных размеров (85–130 нм). Это сравнение показывает, что средний размер синтезированных кристаллитов алмаза D19 находится в области 10 030 ± нм. This estimation coincides well with the representative size observed in the TEM image shown in Fig. 5c. It is notable that the EPR spectrum of D19 recorded at the high power of P MW = 200 mW (Fig. 7a) shows a lack of definition of the HFS structure of the P1 signal. The broad central line suggests the presence, at the nanoscale, of dense clusters of paramagnetic spin-half that strongly couple to each other.
Altogether, the decrease in the linewidth of the main EPR signal (g = 2.0024), the appearance of well-defined HFS characteristics in the P1 centre spectrum after sintering, and the I pp saturation for P MW > 0.7 mW (Fig. 7c) are suggestive of an increase in size by up to one order of magnitude (crystal size > 50 nm). It also indicates a better crystallinity of the nanodiamond in the D19 sample. From van Wyk measurements [29], a smaller amount of paramagnetic defects (< 200 ppm) are expected, based on the narrow linewidth (0.14 mT) of the g = 2.0024 main paramagnetic signal in the D19 sample.
Fluorescence and Raman Scattering of Diamond Crystals
The photoluminescence (PL) spectrum of the DP 0–0.05 precursor together with the PL spectra of some much coarser fractions of polycrystalline diamond particles (DP 0–0.2 and DP 0–0.35) produced by Microdiamant TM is shown in Fig. 8. The spectrum of DP 0–0.05 under the 488 nm excitation wavelength has two features of note:the prominent narrow PL line at 738 nm, associated with the zero-phonon line of negatively charged SiV − centres, and a broad spectrum background with PL bands centred at 525, 600, 660 and 740 nm, associated with various light-emitting centres in diamond, including NV centres. For polycrystals with mean size 25 nm (DP 0–0.05), the intensities of these bands are smaller than that for polycrystals with mean sizes of 100 and 175 nm (DP 0–0.2 and DP 0–0.35, respectively). A more detailed analysis of the PL spectra of polycrystalline DP 0–0.05 particles has been previously undertaken [19].

PL spectra of various submicron fractions of Microdiamant™ polycrystalline diamond particles:blue—DP 0–0.05 (mean size 25 nm), green—DP 0–0.2 (mean size 100 nm), red—DP 0–0.35 (mean size 175 nm). Excitation wavelength λ = 488 nm. The prominent peak at 738 nm marked by the vertical dashed line is the zero-phonon line of negatively charged SiV − centres, which can be observed in all polycrystalline diamond fractions. For better comparison, the spectra are specially normalized for PL intensity at λ = 590 nm. Normalising coefficients are indicated in the figure
Figure 9a shows the PL spectrum of sintered diamond sample D19 (blue curve) at room temperature (RT) together with the PL spectrum of the DP 0–0.05 precursor (red curve). The D19 spectrum displays a green fluorescence characteristic, with a sharp maximum at 525 nm, and a subsequent decrease at larger wavelengths. Note that the single-phonon, sharp Raman line of diamond, which is expected at 522 nm, was too weak to be detected on the ascending slope of the PL signal under 488 nm excitation wavelength. Footnote 6 As previously reported [9, 30], such spectra—with a continuous higher wavelength band of “triangular” shape—are characteristic of optical emission from NVN centres (also known as H3 centres) in submicron (< 140 nm) diamonds at RT. In the PL spectrum of sample D19, at least four broad bands (“bumps”) centred at 538, 569, 601 and 710 nm can be additionally distinguished. We do not believe that they are related to phonon sidebands of the NVN (H3) centres. The origin of the 525 nm sharp peak and “bumps” is still unclear, but it is probably due to an impurity-related complex; the precursor material contains a large number of residual contaminants as mentioned before (see “Structural Characterisation of the Materials Before and After the Sintering” section), some of which are present at significant concentrations (~ 100 ppm). The zero-phonon emission line of NVN at ~ 503 nm wavelength is barely detectable and cannot be distinguished from the two small shoulders (at 500 and 505 nm) in the same region. By comparing the D19 and DP 0–0.05 spectra (Fig. 9a), one can see that they superimpose well for λ > 750 nm. However, the spectra differ significantly in the 480–650 nm range due to the appearance after sintering of NVN centres, which were not present in the precursor material. In order to verify our interpretation of the main optical emission of D19 (in the range 500–650 nm) as originating from NVN centres, we compared the PL spectrum of D19 with the PL spectra of two reference samples (HPHT diamonds, Columbus NanoWorks Inc., US) of two very different sizes and both known to contain NVNs (Fig. 9b). The D19 PL spectrum (dashed line) coincides very well with those of the PL spectra of the HPHT microdiamonds containing NVNs. The emission spectrum of the 100-µm sized reference sample shows a sharp single-phonon diamond Raman line (487.4 nm) and the zero-phonon line (504 nm) of NVN under 457 nm laser excitation at room temperature (Fig. 9b, violet spectrum). The 150-nm sized HPHT nanodiamonds were excited at 488 nm, and it displayed a very similar global photoluminescence spectrum shape as that of the 100-µm sized sample, with a Raman single-phonon line at 522 nm. However, it did not exhibit the NVN zero-phonon line, behaving in that sense exactly like the D19 sample. The “bumps” present in the D19 sample spectrum are absent from both reference PL spectra, indicating that these features are not related to the NVN emission.

PL spectrum of submicron powder D19 sample at T = 293 K (blue line) compared with that of its DP 0–0.05 polycrystalline precursor (red line) under the same conditions with laser excitation at λ = 488 nm (a ), emission spectra, under 457 nm and 488 nm excitation, of two reference synthetic HPHT samples (size ~ 100 μm and < 150 nm) containing NVN centres (b ) and the Raman spectrum of D19 sample recorded using the 457 nm excitation laser radiation (c ). Arrows in (b ):lines at 487.4 nm and 522 nm are single-phonon diamond Raman lines at 457 nm and 488 nm excitation, respectively, and the line at 504 nm is the ZPL of the NVN centres. In (b):dashed line—PL spectrum of D19 at λ = 488 nm excitation (for comparison). In (c ):the diamond Raman line is centred at 1331.4 cm −1 . δ = 7.3 cm −1 is a FWHM of diamond Raman line having the Lorentzian shape
We also measured Raman scattering from D19, in the range 1000–1600 cm −1 , under excitation by 457 nm laser radiation. Figure 9c displays the Raman spectrum, corrected to remove the autofluorescence background. The spectrum consists primarily of a narrow characteristic diamond Raman line centred at 1331.4 cm −1 and an exceptionally broad (width ≈ 100 cm −1 ) band centred at 1450 cm −1 . The latter could be due to non-diamond amorphous carbon phase and/or some transpolyacetylene (TPA) species located at diamond crystals surface [1,2,3]. A further, ill-defined, band at ~ 1090 cm −1 of lower intensity is probably related to TPA species. The broad band at ~ 1450–1480 cm −1 could also be related to multivacancy chains in the diamond lattice and sp 2 - rehybridisation within these chains [6]. Footnote 7 Furthermore, we did not observe the characteristic G-band (centred at 1570–1590 cm −1 ) associated with an sp 2 graphitic nanophase. These observations are indicative that the diamond sample D19 being graphite free, which is also in agreement with its white colour under daylight illumination.
Moreover, the width of the Raman diamond line (7.3 cm −1 ) in the D19 sample is smaller than that for the DP 0–0.05 polycrystalline particles (10.6 cm −1 ). Table 1 contains Raman diamond line data for bead-milled synthetic Ib HPHT diamonds with mean size varying from 25 to 1000 nm (Microdiamant AG, Switzerland). The linewidth decreases from 9.12 to 5.24 cm −1 with increasing size. This can be explained by the lower prevalence of structural defects in larger crystals. Using data as a calibration curve to infer the crystal size from the diamond Raman linewidth of δ ≈ 7.3 cm −1 yields an estimation for the D19 crystal size of ~ 80 nm. This value coincides reasonably with the estimation in “EPR of Polycrystals and Diamond Crystallites Before and After the Sintering” section on the basis of EPR data. Moreover, this value is very similar to our previous published results where diamonds were obtained by HPHT sintering of 5-nm DND in the presence of ethanol [31]. However, the precursor material used for sintered DND and the one used in this work using the smallest (~ 25 nm) fraction of milled Du Pont shock-wave polycrystalline diamonds are considerably different from the viewpoint of crystal types and elementary crystallite sizes. This obtained size is about 8 times larger than the coherent scattering region length of L CSR ≈ 11 nm extracted from XRD earlier, but it is consistent with crystallite having a low density of structural defects, in agreement with the EPR studies.
We also studied the fluorescence from very fine individual D19 particles. For this purpose, the supernatant fraction of diluted and ultrasonicated water suspension of D19 particles obtained after centrifugation at 4500 × g for a 30 min was used. Coarse particles and large loose aggregates with size exceeding ~ 0.2–0.3 micron were absent in such supernatant. Fine D19 particles were spin-coated onto a thin glass coverslip from the diluted supernatant of D19 particles. A typical image of fine fluorescent D19 particles is shown in Fig. 10a in greyscale (mono 14-bit images). It consists of many spots with different brightness. Some spots, such as that marked by the yellow circle, have sizes close to the diffraction limit. The intensity profile of this spot has a Gaussian shape in its central core and a FWHM of about 5–6 pixel corresponding to ~ 440 nm (Fig. 10b). Such spots come from at least quarter-micron particles and particles of smaller size. A greater number of brighter spots correspond to the larger reassembled aggregates of D19 particles having more NVN colour centres and hence the overall emission intensity increasing.

Wide-field fluorescent image of isolated D19 particles obtained by confocal epifluorescence microscopy (a ), and the intensity profile of the selected nanoparticle marked by the yellow circle (b ). Image size: ~ 80 × 80 mμ. One pixel corresponds to 80 nm
Discussion
We showed that under HPHT conditions, and in the presence of ethanol, we can convert polycrystalline diamond particles (composed of tightly cemented nanometre-sized cubic diamond crystallites separated by a non-cubic diamond phase) into larger cubic diamond crystallites. The process probably occurs through recrystallisation of the cubic diamond phase and transformation of non-cubic diamond phases including multiple twin boundaries into diamond. During this process, vacancies appear and can form NVN complexes with nitrogen atom pairs. These complexes have a characteristic photoluminescence in the green. While the EPR spectra of the precursor polycrystalline diamonds show NV − triplet centres, triplet multivacancies, and SiV − centres, none of these were present after the sintering. The disappearance of NV − centres and multivacancies has previously been observed [31] after sintering detonation nanodiamonds at HPHT conditions (P = 7 GPa and T ≥ 1350 °C) with ethanol. Footnote 8 The presence of multivacancies is a characteristic feature of damaged diamond lattices, with defects mainly located in a thin layer of ~ 2 nm at the surface. The absence of the paramagnetic triplet and SiV colour centres is strong evidence that substantial recrystallisation took place, accompanied with the appearance of new defect types (NVN). The saturation trend of the substitutional nitrogen (P1 centre) EPR signal with increasing microwave power indicates long spin–spin and spin–lattice relaxation times. These are signatures of improvement of the quality of diamond crystal lattice after sintering. We can assume that during HPHT sintering, vacancies from empty spaces within or between polycrystals join with A-centres to form the new NVN entities.
Выводы
Sintering of diamond polycrystals, with size varying from 3 to 50 nm, in the presence of ethanol, lead to the substantial enlargement of elementary diamond nanocrystals and improved their crystalline quality. During this process, SiV − and NV − colour centres present in the precursor nanodiamond disappeared, while the EPR signature of P1 substitutional nitrogen paramagnetic centres appeared. We also observed the green photoluminescence of NVN colour centres. The comparison of the FWHM of diamond Raman line (~ 1332 cm −1 ) of the synthesised selected microcrystals under study with those of a series of reference samples revealed that the mean size of diamond crystals after sintering is approximately 80 nm. The analysis of the EPR spectrum dependence upon microwave power demonstrated the good crystalline quality of the synthesised sintered diamond with a concentration of P1 centres smaller than 200 ppm. Hence, our technique of HPHT sintering is a strong alternative to conventional high-energy particle beam irradiation [9] to form NVN centres in nanodiamond. It can be used to produce purely “green” fluorescing nanodiamonds with no (or very limited) crosstalk with the “red” fluorescing nanodiamonds (containing NV 0 and NV − centres), as required in biolabelling for cathodoluminescence integrated correlation electron-light microscopy [32].
Доступность данных и материалов
The data underpinning this manuscript is available from the corresponding author on request.
Notes
- 1.
The apparatus response function of the XRD diffractometer was determined from a LaB6 reference sample.
- 2.
One mechanism explaining the lack of HFS in the EPR signal of paramagnetic nitrogen in size < 50 nm diamond nanoparticles is described in detail elsewhere [21]. Alternative mechanisms are also not excluded [33, 34].
- 3.
X-ray diffraction CSR size is ~ 4 times smaller in this case.
- 4.
This narrow EPR signal is slightly asymmetrical and consists of at least two components related to P1 centres and to defects having C–C dangling bond spins S = 1/2. The intensity of the second component represents at least 30% of the intensity of the main peak.
- 5.
Here we assume the sequence of untreated first-derivative experimental EPR spectra measured at different P MW , two of which are shown in Fig. 7a.
- 6.
However, the Raman line was detected at 457 nm excitation.
- 7.
The Raman band centred at ~ 1480 cm −1 was found for HPHT diamonds grown with magnesium-based catalysts [6]. Such diamond crystals may contain SiV and GeV colour centres.
- 8.
Instead of a multivacancy g = 4.00 singlet EPR signal, a new EPR signal with a quintet hyperfine structure related to N⋯N pairs separated by no more than 0.7 nm was observed in this case.
Сокращения
- NV − :
-
Вакансия азота
- NVN:
-
Nitrogen-vacancy-nitrogen
- HPHT:
-
High-pressure, high-temperature
- DND:
-
Detonation nanodiamond
- EPR:
-
Electron paramagnetic resonance
- FWHM:
-
Полная ширина на половине максимальной
- XRD:
-
Рентгеновская дифракция
- LCSR :
-
Coherent scattering region length
- HRTEM:
-
Просвечивающая электронная микроскопия высокого разрешения
- PL:
-
Фотолюминесценция
- MW:
-
Microwave
- HFS:
-
Hyper-fine structure
- RT:
-
Комнатная температура
Наноматериалы
- Что такое алмазная токарная обработка?
- Что такое селективное лазерное спекание?
- Бриллиант
- Наноалмазы для магнитных датчиков
- Влияние небольшого количества SiO2 на кинетику спекания нанопорошков тетрагонального диоксида циркония
- Моделирование молекулярной динамики и имитация алмазной резки церия
- Стратегия гидротермального спекания для анодного материала LiNb3O8 с пористой и полой структурой
- Матрицы нанополосов на основе GaAs с золотым покрытием, изготовленные методом химического травления с примене…
- Поведение при спекании SiC, спеченного плазменной искрой, с композитными наночастицами Si-SiC, полученными метод…
- Перовскитные солнечные элементы, изготовленные с использованием экологически чистой апротонной полярной до…