


Фотолюминесценция от NV- центров в детонационных наноалмазах 5 нм:идентификация и высокая чувствительность к магнитному полю
Аннотация
Содержание азотных вакансий (NV - ) центров окраски в наноалмазах (ДНА), образующихся при детонации азотсодержащих взрывчатых веществ, составляет 1,1 ± 0,3 ppm. Это значение впечатляет для наноалмазов размером <10 нм с намеренно созданным NV - центры. Концентрация оценивалась с помощью электронного парамагнитного резонанса, определяемого по интегральной интенсивности g =4,27 строка. Эта линия связана с «запрещенным» ∆ m s =2 перехода между уровнями Зеемана NV - состояние основного триплета центра. Конфокальная флуоресцентная микроскопия позволяет обнаруживать красную фотолюминесценцию (ФЛ) NV - центры окраски в наноразмерных агрегатах ДНА, образованных из наночастиц размером 5 нм. Субволновые излучатели, состоящие из NV - с размерами в несколько раз меньшими, чем дифракционно ограниченное пятно. Далее мы наблюдали резкое падение интенсивности ФЛ при смешивании и антипересечении спиновых уровней основного и возбужденного состояний в NV - происходит под действием приложенного внешнего магнитного поля. Этот эффект является уникальной квантовой особенностью NV - центры, которые нельзя наблюдать для других светоизлучающих центров окраски видимой области в решетке алмаза.
Фон
Флуоресцентные наноалмазы (НА), содержащие вакансию азота (NV - ) центры представляют собой новые наноматериалы, которые открывают путь к инновационным приложениям. Конкретные приложения в настоящее время включают магнитное зондирование [1], биоимиджинг [2], а также телекоммуникации и обработку информации, включая использование источников фотонов, связанных с нанорезонатором [3, 4]. Широкий спектр областей применения проистекает из беспрецедентных уникальных квантовых свойств NV - центры окраски, возникающие при комнатной температуре. Центры окраски обладают триплетными спиновыми свойствами, которые могут быть обнаружены с помощью оптически детектируемого магнитного резонанса (ODMR) [5]. Флуоресцентные свойства наноалмазов в сочетании с их небольшими размерами (<40 нм) позволяют использовать их в биомедицинских приложениях, включая внутриклеточную контрастную визуализацию субмикронных органелл и термометрию в нанометровом масштабе у эмбрионов [6,7,8]. Инкапсуляция кристаллитов наноалмаза в биосовместимые полупрозрачные оболочки особенно перспективна для их применения в биологических средах [9]. Их также можно использовать в качестве точечных датчиков для измерения шума Джонсона в металлах [10]. Наноалмазы могут образовываться во время детонации взрывчатых веществ, давая так называемые детонационные наноалмазы (ДНА). Однако ДНА обычно содержат высокую концентрацию недостатков. Это ограничивает применение, например, ДНА (размер <6 нм) в приложениях магнитного зондирования и других локальных исследованиях на наноуровне [11].
ДНА синтезируется из продуктов пиролиза энергосберегающих компонентов ВВ при детонации. Взрывчатое вещество и детонация содержатся в резервуаре с водой. Синтез происходит за время, например, 13–20 мкс при детонации блока ВВ размером до 10 см. Это время, необходимое для распространения детонационной волны через весь блок взрывчатого вещества. Однако каждая отдельная микрогранула взрывчатого вещества разлагается и превращается в газ за гораздо более короткое время (<0,1 мкс). Углерод и азот являются преобладающими компонентами взрывчатых веществ. В процессе детонации алмазные кристаллиты размером до 5 нм самоорганизуются из продуктов разложения компонентов взрывчатого вещества, то есть преимущественно углерода. Азот также включается в решетку алмаза во время синтеза. Наличие CH 3 * радикалы в продуктах газофазного разложения вызывают быструю сборку решетки алмаза в сложной газовой среде C – N – O – H. Газовая среда изменяется в микросекундном масштабе, с быстрой разгрузкой давления и перепадами температуры, создавая условия окружающей среды, соответствующие требуемой области для синтеза алмаза и стабильности на Р – Т-фазовой диаграмме углерода [12].
Чрезвычайно быстрая сборка алмазных кристаллитов в центрах конденсации (а также неалмазных форм аморфного углерода) вызывает появление большого количества дефектов, вакансий и мультивакансий априори разной морфологии внутри растущей решетки. Вакансии естественным образом возникают в результате дефектов упаковки и неточностей, возникающих во время быстрой сборки алмазной решетки. При понижении температуры от максимума при детонации до 800–900 ° C вакансии еще могут перемещаться внутри решетки. Таким образом, вакансии могут объединяться в кластеры или исчезать на поверхности кристаллитов. Подвижность вакансий на этой стадии синтеза также позволяет захватывать их замещающими атомами азота. Те же процессы применимы и к примесям азота, с той лишь разницей, что изолированные азотные замещения практически неподвижны в решетке при температурах ниже 1600 ° C и, следовательно, агрегируются только при высоких температурах. Примеси азота могут присутствовать в решетке ДНА в различных формах. Он может присутствовать в агрегированной форме (А-центры, димеры) в количестве до ~ 2,5 ат.% Или в виде изолированных углеродзамещенных атомов (С-центры). Как известно, A-центры непарамагнитны и имеют нулевой спин, тогда как C-центры с половиной спина являются парамагнитными в нейтральной форме (P1-центры). Следовательно, P1-центры могут быть легко обнаружены методом электронного парамагнитного резонанса (ЭПР). Вакансия в решетке, будучи отрицательно заряженной (V - ), также парамагнитен, но имеет спин 3/2. Спин-полужирные спины были обнаружены в ДНА в преобладающем количестве [13]. Концентрация спин-полурадикалов, обнаруженная методом ЭПР, практически одинакова (1100–1400 ppm) Footnote 1 во всех типовых ДНА промышленного производства. Это не зависит от конкретных коммерческих производителей и деталей их технологии, которые могут отличаться более мелкими деталями. Концентрация спин-половинного радикала приблизительно равна сумме общего числа замещающих атомов азота и других дефектов, имеющих оборванные спины связи внутри частицы ДНА, по сравнению с массой частицы (или соответствующим количеством атомов углерода в ней). Большая часть азота (до ~ 2,5 ат.%) В ДНА распределена более или менее однородно в ковалентной решетке алмазных ядер, но обогащена в дефектных областях, таких как дефекты упаковки [14, 15]. К такому выводу пришли Тернер и др. что внедренный азот преимущественно присутствует в решетке алмаза в форме N – N димеров (A-центров) или изолированного нейтрального, положительно или отрицательно заряженного азота sp 3 -координированные примеси (С-центры) [14, 15]. Таким образом, большая часть азота в ДНА находится в непарамагнитной форме пар азота (N – N), расположенных в соседних узлах кристаллической решетки алмаза.
Помимо преобладающих примесей азота, частицы ДНА также содержат изолированные вакансии, поливакансии, дефектные центры типа азот-вакансия-азот (NVN) и азот-вакансия (NV). Эти дополнительные дефектные центры возникают из-за спонтанного появления составляющих агентов в соседних узлах решетки во время сборки решетки [11]. В то же время наличие отрицательно заряженных НВ - Это происходит в основном из-за их зарядки и принятия отрицательного заряда от избыточных нейтральных атомов азота, играющих роль доноров электронов в системе. По нашим предварительным данным ЭПР, концентрация NV - центров, которые можно определить по интегральной интенсивности линии ЭПР с a g -фактор 4,26–4,27 в половинном магнитном поле примерно на 2,5–3 порядка меньше, чем концентрация изолированного замещающего азота, что дает значение 400–700 ppm.
Мультивакансии и NV - Центры были успешно идентифицированы в ДНА методом ЭПР в нашей предыдущей недавней работе [13, 16, 17]. Также были предприняты попытки понять взаимосвязь между яркостью NV - центры в ДНА, химический состав их поверхности, окружение и морфология агрегатов [18, 19]. Аналогичные работы по контролю эмиссионных свойств NV - также было сделано для синтетических флуоресцентных алмазов с искусственно созданными NV-центрами [20]. Тем не менее, необходимые исследования, сочетающие в себе различные методы анализа, включая порошковую рентгеновскую дифракцию, спектроскопию ЭПР, конфокальную флуоресцентную спектроскопию и микроскопию, элементный анализ, анализ излучателей дифракционно ограниченных размеров и влияние магнитного поля на сигнал фотолюминесценции, полностью не выполнены. по теме НВ - Центры в ДНА, за исключением нашей предварительной работы [21]. В настоящее время зависимость эмиссионных свойств НВ - в ДНА не изучены внутренние примеси и морфология частиц. В этой работе мы сообщаем, что частицы ДНА демонстрируют специфическую фотолюминесценцию (ФЛ) NV - центры, точно подсчитать содержимое NV - в них с помощью сложного подхода и проанализировать их интенсивность ФЛ в максимуме спектра в зависимости от размера кристаллитов алмаза и содержания азота. Кроме того, мы демонстрируем чувствительность ДНА к внешнему магнитному полю, которая связана с их свойствами ODMR.
Экспериментальный
Методы изготовления и управление морфологией DND
Порошки ДНА были получены с помощью в основном стандартного процесса, который включает подрыв около 1 кг взрывоопасной смеси тринитротолуол-гексоген, окруженной водной оболочкой соответствующей толщины, в закрытом сосуде из нержавеющей стали объемом несколько кубических метров. Это так называемый «мокрый» синтез. Синтез и первичная очистка продукта проводились коммерческим производителем - СКБ «СКТБ Технолог», г. Санкт-Петербург, Россия. Технологический процесс, применяемый на предприятии, позволяет получать порошки ДНА, состоящие исключительно из нанокристаллитов с размером области когерентного рассеяния (ОКР) от 4,5 нм до примерно 5,7 нм. Часть очищенных порошков ДНА, приготовленных по стандартной технологии, была поставлена компанией PlasmaChem GmbH (Берлин, Германия). Морфология доставленных образцов ДНА была установлена методом рентгеновской дифракции (XRD). Рентгенограммы регистрировали в Центре исследований микро- и нанотехнологий Университета Хосей (Токио, Япония) с помощью рентгеновского дифрактометра Rigaku SmartLab I с детектором D / teX Ultra, источником излучения CuKα ( λ =1,54178 Å) и никелевый фильтр в диапазоне углов 2Θ =5–100 o (шаг 0,01 o ). При анализе учитывалась ширина аппаратной функции. Рентгеновские размеры ОКР были определены на основе сложного анализа ширины рефлексов 111, 220 и 311 как функции угла 2Θ в соответствии с методом, описанным в [4]. [22]. Сноска 2 Ключевой особенностью этого метода является то, что параметры постоянной решетки ( a о =3,5640 Å) можно получить для всех образцов ДНА с систематическими ошибками (вариациями из-за выбора разных моделей) не более ± 0,0003 Å. Измерения дифракции рентгеновских лучей на порошке показали, что размер ОКР одного репрезентативного образца ДНА, выбранного для последующих исследований, составляет 5,2 ± 0,2 нм. Этот образец ДНА использовался во всех основных исследованиях, описанных ниже. Кроме того, для построения зависимости интенсивности фотолюминесценции от размера ОКР и / или содержание внутреннего азота. Размер ОКР порошка ДНА, синтезированного по обычной технологии, составил 4,6 ± 0,2 нм.
Порошки ДНА были дополнительно очищены в смесях кипящих кислот для удаления любых остаточных примесей 3d-ферромагнитных металлов, что позволило провести более точные исследования методом ЭПР и РФЭС. Для исследования комбинационного рассеяния света и последующих исследований фотолюминесценции требовалась дополнительная очистка поверхности путем отжига порошков ДНА на воздухе при 430 ° C в течение 10–12 ч [23]. Никаких дополнительных усилий по деагрегированию порошка ДНА, содержащего отдельные дискретные частицы наноалмаза и агрегаты с размерами менее 25 нм, после кислотной очистки и обработки на воздухе не предпринималось. Средний размер агрегатов ДНА в водной суспензии составлял 25–30 нм.
Образцы для конфокальной флуоресцентной микроскопии были изготовлены из водных суспензий ДНА. Суспензии получали диспергированием очищенного кислотой и отожженного на воздухе порошка в воде с концентрацией ≈ 1 мг / мл с последующим разбавлением водой в 100 раз. Затем суспензии ДНА наносили центрифугированием на слой 170 мкм. -толстая покровная стеклянная подложка, предварительно очищенная в смеси этанола и ацетона в ультразвуковой ванне. Наконец, агрегаты ДНА, осажденные на подложках из покровного стекла, очищали УФ / О-очистителем (ртутная лампа низкого давления:оптическая мощность> 25 мВт / см 2 при λ =254 нм) примерно на 30 мин. Под воздействием ультрафиолетового излучения происходит эффективное окисление и газофазное травление поглощающих свет sp 2 - образуется углерод вокруг ДНА с кислородом и озоном. Внешние наноразмерные кластеры sp 2 -углероды крайне нежелательны для изучения ФЛ от собственных центров окраски в решетке алмаза ДНА. Воздействие УФ-излучения на подложку также приводит к удалению любых нежелательных органических загрязнителей, которые в противном случае могут вызвать люминесцентный фон в широком спектральном диапазоне.
Методы характеризации
Магнитно-резонансная спектроскопия
Спектры ЭПР образцов регистрировали при комнатной температуре в микроволновом Х-диапазоне с частотой ~ 9,4393 ГГц или ~ 9,0785 ГГц с использованием спектрометра ЭПР (JEOL JES-FA 300 (Япония)). Конкретная используемая фиксированная частота зависела от наличия или отсутствия кварцевого адиабатического криостата в микроволновом резонаторе в течение всего цикла исследования. Десятки миллиграммов порошка ДНА были введены в длинную X-полосную трубку JEOL EPR (JEOL Parts Catalog, item 4220 00281, № 193) с внешним диаметром 5 мм и длиной 100 мм кварцевой нижней стороной. Открытый верх трубки был закрыт от влаги. Фотографии порошка ДНА в ЭПР-трубке JEOL и в центре СВЧ-резонатора показаны на рис. 1 а и б. Порошок серого цвета хорошо виден на обеих панелях. Такое количество порошка ДНА подходит как для эффективной настройки спектрометра ЭПР с высокой добротностью, так и для получения хорошего отношения сигнал / шум при регистрации спектров слабых сигналов в области половинного магнитного поля.

Фотографии порошка ДНА в а трубка JEOL EPR и b в центре СВЧ-резонатора X-диапазона. Для сравнения изображение порошка в пробирке для ЭПР специально снято на фоне основной части пластикового шприца объемом 10 мл
Спектры ЭПР ДНА с g -факторы сигналов в диапазоне 4.00–4.30, зарегистрированные при мощности СВЧ, P МВт =6 мВт; амплитуда модуляции магнитного поля, А м =0,5 мТл; частота, ν =100 кГц; усиление, G =1200; и накопление сигнала, N =20. Постоянная времени 0,030 с. Время записи одной развертки магнитного поля составляло 120 с.
Минимальные и максимальные значения магнитного поля для развертки были специально выбраны с помощью программного обеспечения JEOL-JES и точно определены с помощью измерителя поля ЯМР JEOL ES-FC5 (Echo Electronics). Концентрация НВ - центров оценивали путем двойного интегрирования соответствующего сигнала ЭПР с g =4,27, с последующим присвоением соответствующей массе образца и сравнением с сигналом ЭПР от эталонного образца. Мы специально выбрали синтетический флуоресцентный алмазный порошок Ib HPHT Footnote 3 (средний размер ~ 100 нм) с хорошо выраженной интегральной интенсивностью g =4,27 линия ЭПР и известная концентрация S =1 НВ - центров 5,3 ± 0,4 ppm в качестве независимого надежного эталона, позволяющего правильно определить полученную концентрацию NV - в режиме "Не беспокоить". Это похоже на подход, предложенный ранее Shames et al. [24]. g =4,27 линия ЭПР была записана для этого эталонного образца со следующими параметрами: P МВт =3 мкВт, А м =0,2 мТл, G =500 и N =12 в линейном режиме, где размах линии ЭПР пропорционален квадратному корню из микроволновой мощности в пространстве образца. Для кристаллов Ib HPHT субмикронных и микронных размеров этот режим обычно хорошо реализуется при низких мощностях микроволн (ниже 7–10 мкВт). Спектр был специально записан в широком диапазоне магнитных полей от 119 до 321 мТл. Этот диапазон использовался для регистрации дополнительных сигналов ЭПР от Δ м s =1 переходы, которые разрешены с низким полем, и представляют уникальные сигнатуры NV - центров.
Рентгеновская фотоэлектронная спектроскопия
Рентгеновскую фотоэлектронную спектроскопию (XPS) ДНА проводили с использованием системы Perkin-Elmer PHI 5600 Multi-Technique, оснащенной источником монохроматического излучения Al Kα. Спектральные характеристики фотоэлектронов были получены после анализа потока фотоэлектронов, испускаемого из образца площадью ~ 1 мм 2 . [25]. Обнаруженные фотоэлектроны испускаются с поверхности материала - нескольких областей толщиной в несколько атомных слоев (<2 нм). Особое внимание было уделено анализу площадей фотоэмиссионных пиков углерода C1s, азота N1s и кислорода O1s. Разложение пиков фотоэмиссии на отдельные компоненты производилось по соответствующему набору синглетных контуров. Спектры образца ДНА снимались после травления поверхности ионами аргона. Ионное травление проводили в течение 3 мин при ускоряющем напряжении 3 кВ и токе ионного пучка около 4 мкА. Травление ионами Ar использовалось для удаления адсорбированных окружающих газов и других хемосорбированных атомных комплексов с поверхности образца. XPS также использовался для оценки внутреннего содержания азота в серии различных образцов ДНА с разным размером CSR на рентгеновских лучах.
Флуоресцентная спектроскопия
Для исследования ФЛ образцы готовили путем вдавливания порошка заподлицо в неглубокое отверстие диаметром 2 мм, выполненное в медной пластине 4 мм, с последующим выравниванием поверхности стеклянной пластиной. Спектры ФЛ регистрировались в диапазоне 540–1000 нм с помощью нестандартной установки микро-комбинационного рассеяния света с возбуждением лазером на длине волны 532 нм и мощностью ~ 0,5 мВт и дифракционной решеткой с 600 штрихами на миллиметр. Время экспозиции 180 с. Возбуждающее излучение фокусировалось на плоской поверхности образца через объектив микроскопа Nikon Plan Apo 100 ×, NA =1,40 в пятно диаметром 2 мкм. Спектральное разрешение было лучше 0,05 нм.
Конфокальная флуоресцентная микроскопия
Для регистрации спектров ФЛ и картирования сигнала ФЛ от частиц ДНА, нанесенных методом центрифугирования на покровное стекло, использовалась дополнительная установка ФЛ. Он состоит из настраиваемого инвертированного конфокального сканирующего микроскопа в сочетании со спектрографом для визуализации. Конфокальный микроскоп включает пьезоэлектрический сканирующий столик (NanoPDQ75, Mad City Labs Inc., США), объектив микроскопа (Plan Apo 100 ×, NA =1,40, Nikon, Япония) и полупроводниковый лазер с оптической накачкой (Verdi, Coherent Inc., США), излучающий на длине волны 532 нм, который вводится (круговая поляризация, мощность 0,5 мВт) в объектив через дихроичный светоделитель (z532rdc, Semrock, США). Тракт обнаружения состоит из точечного отверстия диаметром 50 мкм для конфокального подавления, полосового фильтра (FF01-697 / 75, Semrock) и детектора модуля счета одиночных фотонов (SPCM-AQR-14, Perkin-Elmer, Канада). . Спектрограф для визуализации состоит из вогнутой решетки, отображающей спектр на ПЗС-матричном детекторе с задней подсветкой (DU440-BU2, Andor Technologies, UK), что обеспечивает спектральное разрешение около 1 нм.
Картирование сигнала люминесценции производилось на 20 × 20 мкм 2 квадратный. Смещение элементарной ступени в поперечном направлении составляло 100 нм с точностью ~ 1 нм. Для получения спектров ФЛ в диапазоне 500–900 нм и зависимости интенсивности ФЛ от времени в максимуме соответствующего спектра были выбраны два из нескольких агрегатов ДНА, обнаруженных на подложке с латеральными размерами не более 0,6 мкм и обладающих интенсивной люминесценцией. . Время экспозиции составляло 60 с для каждого спектра ФЛ и 3 мс на пиксель в каждой зарегистрированной карте 2D-ФЛ. Размер пятна, ограниченный дифракцией (функция рассеяния точки, PSF), составляет около 2–3 пикселей (250 нм). Магнитное поле прикладывалось с помощью постоянного магнита, расположенного на расстоянии примерно 1 мм от подложки из покровного стекла. Напряженность магнитного поля в месте нахождения образца составляла около 90–100 мТл. Зависимость сигнала ФЛ от времени регистрировалась при многократном быстром приближении и удалении магнита от поверхности с образцом и от нее.
Результаты и обсуждение
Подсчет NV - Центры в ДНА методом полуполевого ЭПР
Поскольку излучение центров окраски может поглощаться sp 2 -содержащие частицы, присутствующие внутри агрегатов ДНА и даже могут быть заблокированы определенными морфологическими особенностями в пространстве между частицами, очень важно контролировать и контролировать NV - центры в алмазах неоптическими методами. NV - центры могут быть обнаружены с помощью спектроскопии ЭПР даже при встраивании их носителей в непрозрачную плотную среду, которая блокирует как возбуждающее, так и люминесцентное оптическое излучение, или в случае отсутствия NV - вторичное излучение, связанное со специфическими механизмами тушения. Их общую концентрацию впоследствии легко оценить, анализируя спектр ЭПР [16, 26, 27]. При этом определяемая концентрация НВ - центры предоставляют ценную информацию о потенциальной (после обширной очистки) яркости NV - PL. Спектр ЭПР порошка ДНА, очищенного от магнитных примесей 3d переходных металлов (в основном Fe, Ni) в области так называемого половинного магнитного поля, показан на рис. 2 (черная кривая). Спектр состоит из двух близких линий (1 и 2) с g -factors g 1 =4,27 и г 2 =4,00, а ширина ΔH pp1 ≈ 2,0 мТл и ΔH pp2 ≈ 1,4 мТл. Первая слабопольная линия соответствует запрещенным переходам Δ m s =2 между зеемановскими уровнями энергии триплетного состояния НВ - центр в магнитном поле. Вторая, сильнопольная линия, соответствует тем же запрещенным переходам Δ m s =2, которые возникают, когда микроволновое излучение поглощается другими центрами, т.е. мультивакансиями, имеющими S =1. В исх. [16] эти линии были впервые отнесены к разным триплетным центрам, сосуществующим в ДНА, и, в частности, к g 1 =4.27 строка была назначена NV - центр. Положение g 1 =4.27 соответствует линии запрещенных переходов Δ m s =2, как показано на энергетической диаграмме основного состояния синглет-триплета NV - центр в магнитных полях до 300 мТл (рис. 3). Большой параметр расщепления спин-гамильтониана в нулевом поле ( D =850 × 10 −4 см −1 ), обусловленное сильным обменным взаимодействием составляющих спинов в триплете NV - по центру, обуславливает высокую чувствительность положения g 1 =4,27 (сдвиг на 0,5% на g -масштаб) к конкретной микроволновой частоте в диапазоне от 9,0 до 9,9 ГГц (X-диапазон) [26]. Интегральная интенсивность линии g 1 =4,27 можно использовать для точной оценки концентрации NV - центров, даже когда люминесценция наноалмазов не обнаруживается из-за поглощения или тушения из-за соседних оптически активных частиц или даже маскирования сильной паразитной фоновой люминесценцией от третьих сосуществующих частиц. Облученный высокоэнергетическими электронами синтетический субмикронный алмаз (средний размер 100 нм) с известной концентрацией NV - центров использовали в качестве эталонного образца (образец Ib HPHT FND). Спектр ЭПР образца Ib HPHT FND, полученный при низкой мощности микроволн P МВт =3 мкВт для сравнения показан на рис. 2 (красная кривая). В диапазоне ниже 320 мТл он состоит из трех четко определенных сигналов ЭПР, соответствующих «запрещенному» Δ m s =2 (при 157,85 мТл) и два допустимых z , Δ м s =1 и x , y , Δ м s =1 (при 234,39 мТл и 281,27 мТл) переходов между зеемановскими уровнями энергии основных триплетных состояний NV - в магнитном поле. Δ м s =2 и z , Δ м s =1 также показаны на схеме рис. 3. Достаточно удивительно, что по сравнению с субмикронным эталонным образцом с NV - центров частицы ДНА демонстрируют только g =4,27 линия ЭПР, связанная с Δ м s =2 «запрещенных» перехода. Примечательно, что допустимые z , Δ м s =1 и x , y , Δ м s =1 микроволновых переходов не наблюдается. Вероятное объяснение - аномальное уширение линий ЭПР, связанное с Δ m s =1 из-за большого изменения основных параметров спин-гамильтониана ( D и E ) для S =1 НВ - по ансамблю частиц ДНА. Таким образом, полное отсутствие строк, связанных с допустимым z , Δ м s =1 и x, y, Δ м s =1 переходы - ключевая нестандартная особенность NV - в DND. По данным коммерческого поставщика, ожидаемая концентрация NV - в эталонном Ib HPHT FND, оцененная на основе поглощенной дозы облучения, составила ~ 5,3 ppm. Интегральная интенсивность g 1 =4,27 в спектре ЭПР ДНА оказалось в ~ 4,8 раза меньше, чем в спектре ЭПР Ib HPHT FND, и, следовательно, концентрация NV - центров в ДНА составляет 1,1 ± 0,3 ppm. Соответствующие процедуры интегрирования обоих сигналов ЭПР в диапазоне половин магнитного поля (130–180 мТл) вместе с поправкой на фон специально показаны на рис. 4. На рис. 4а показан исходный, измеренный спектр ЭПР первой производной. микроволнового поглощения до математической обработки (кривая 1); тот же спектр ЭПР после интегрирования (верхняя кривая 2); и такой же, как измеренный исходный спектр ЭПР, но после вычитания широкой линии лоренцевского типа, связанной с оставшимися железосодержащими комплексами (нижняя кривая 3). На рис. 4b показаны спектры ЭПР как эталонного образца (верхняя кривая), так и ДНА (нижняя кривая) после интегрирования измеренных спектров ЭПР с поправкой на фон. Истинный интегральный спектр самого ДНА (рис. 4b, нижняя кривая) сильно отличается от второго спектра на рис. 4а, где был интегрирован только нескорректированный, измеренный спектр ЭПР ДНА. Полученный спектр (рис. 4б, нижняя кривая) демонстрирует очень глубокую впадину между соседними сигналами ЭПР ( g 1 =4,00 и г 2 =4,27) лоренцевского типа на расстоянии ~ 10 мТл. Заштрихованные области под спектрами ЭПР, показанными на панели (b) рис. 4, показывают интегральные интенсивности линии g 1 =4,27 для обеих сравниваемых выборок. Соотношение этих площадей после нормализации по весу выборки составляет ~ 4,8. Интересно, что для ДНА форма линии g 1 =4,27 является симметричным и широким. Эта линия частично перекрывается высоким полем g 2 =4,00 линия EPR, вызванная мультивакансиями. Некоторый оставшийся нескорректированный фон в виде сильной базовой линии (в основном из неизвлекаемых железосодержащих комплексов) отчетливо виден на рис. 4а (спектр 2). Таким образом, правильная декомпозиция спектра ЭПР ДНА по крайней мере для трех перекрывающихся компонентов в диапазоне 130–180 мТл является первоочередной необходимостью для правильной оценки интегральной интенсивности g =4,27 линия и концентрация НВ - . Мы также обнаружили, что различные обработки при температурах ниже 500 ° C (в вакууме и на воздухе) практически не влияют на концентрацию NV - центры в ДНА. Двойная интегральная интенсивность сигнала g 1 =4,27 (область тени ниже лоренцевского контура с центром ~ 151 мТл для ν =9,0785 ГГц) остается практически таким же после обработки, как и раньше. Это означает, что NV - центры расположены достаточно глубоко в решетке, на глубине от поверхности наночастиц, по крайней мере, с постоянной решетки (~ 0,36 нм), поэтому они не могут реагировать с внешними химическими агентами, которые не травят или не проникают в алмазную фазу.
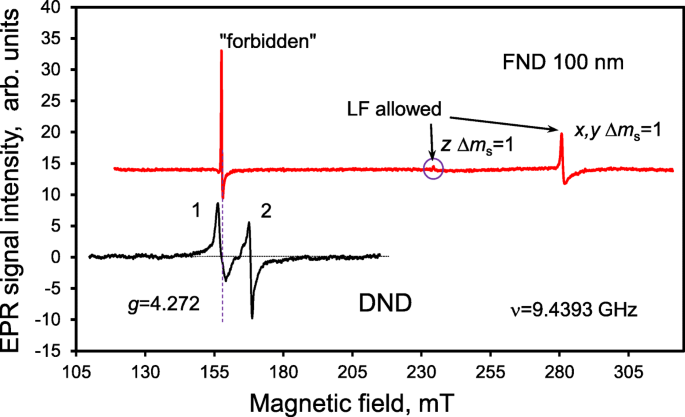
Спектры ЭПР кислотно-очищенных ДНА в области половинного магнитного поля (черная кривая) и эталонных электронно-облученных флуоресцентных наночастиц алмаза Ib HPHT со средним размером ~ 100 нм (красная кривая) в диапазоне до 320 мТл. Обе линии спектра ЭПР ДНА отмечены цифрами 1 и 2. Низкопольная линия 1 значком g =4,272 соответствует NV - центры. Допускаются строки, относящиеся к низкому полю (LF) z Δ м s =1 и x , y Δ м s =1 переходы в FND 100 нм отмечены стрелками для верхнего спектра. Самая слабая, едва различимая линия, связанная с допустимым z Δ м s =1 переход дополнительно отмечен кружком. Микроволновая частота составляла 9,4393 ГГц
.
Energy diagram of ground singlet-triplet levels 3 A2 of NV − in magnetic field up to 300 mT. “Forbidden” Δm s = 2 and LF allowed Δm s = 1 transitions caused by absorption of microwave radiation (ν ≈ 9.44 GHz) are marked by vertical red arrows. The position of the ground state spin level anti-crossing (|0 〉GS and |−1〉GS ) is marked by a circle

а Background subtraction in EPR signal of DND and b estimations of the double-integrated intensities of the g = 4.27 line for a DND sample and reference fluorescent Ib HPHT synthetic diamond. Панель a :as-registered first derivative EPR signal of the DND in the ± 15 mT half magnetic field range (curve 1, blue); the same, but integrated EPR signal in the same ± 15 mT range of magnetic field (curve 2); the same first derivative EPR signal of DND, but after subtraction of the broad parasitic EPR signal from remaining non-removable iron-containing paramagnetic complexes shown by red contour of Lorentzian shape in the upper curve (curve 3, blue). Панель b :Estimation of the double integrated intensity of the g = 4.27 line for a fluorescent Ib HPHT diamond having NV − (upper curve, shaded area) and the DND sample (bottom curve, shadow area). The bottom spectrum in panel b consists of two contours of Lorentzian shape, one of which centred at lower magnetic field (≈ 150.932 mT), is assigned to the NV − centres of DND (the area below this contour in red is highlighted). Microwave frequency:ν = 9.0785 GHz
The main high-intensity EPR signal of the DND lies above 320 mT at ν = 9.4393 GHz and has a g -factor g ≈ 2.0027. It has a Lorentzian curve profile of linewidth ΔH pp = 0.84 mT [13]. This linewidth is greater than that of fluorescent nanodiamonds (FNDs, 100 nm) milled from microcrystalline diamond synthesized by a high-pressure and high-temperature method. The broad signal can be explained by greater exchange and dipole-dipole interactions between the S = 1/2 spins in the spin ensemble within an individual DND nanoparticle than those within a FND. The intensity of the main EPR signal collected from all S = 1/2 paramagnetic centres, both of nitrogen (P1 centres [28]) and non-nitrogen origin, indicates a spin concentration of ~ 1300 ppm, corresponding Footnote 4 to around 15 S = 1/2 spins in each DND [13]. However, it can be concluded from the earlier obtained data that approximately 40–50% of the total number of all paramagnetic centres in the DNDs are due to half-populated antibonding orbitals of isolated P1 centres. Thus, the huge total of spin-half, point-like agents located in DNDs (N ПК ) can be represented as the sum of at least two contributions, from P1 centres and from elemental point defects having dangling bond spins ½, for example based on vacancies like H1 centres (VH o ):\( \left({N}_{\mathrm{PC}}=\left[{\mathrm{N}}_{\mathrm{s}}^{\mathrm{o}}\right]+\left[\mathrm{et}\ \mathrm{al}\ \right]\ \right) \). Here we assume that specific EPR signatures from the hyperfine structures of P1 and H1 centres are absent or greatly “smeared” through the dense arrangement of localized spins within each particle. This sum gives us a clue about the approximate content of both isolated nitrogen and monovacancies inside the DND, although isolated nitrogen and monovacancies can also be present in the diamond lattice in nonparamagnetic forms such as \( {\mathrm{N}}_{\mathrm{s}}^{-},{\mathrm{N}}_{\mathrm{s}}^{+} \), V o . The presence of H1 centres together with neutral monovacancies in DND at some minor level (< 700 ppm) is reasonable in principle because rapid assembling of diamond lattice during the detonation takes place from hydrogen-containing products of TNT-hexogen pyrolysis such as CH3 * or CH2 * radicals. Although the mutual charge transfers between the main groups of centres present in the lattice in various charge and spin states (\( {\mathrm{N}}_{\mathrm{s}}^{\mathrm{o}},{\mathrm{N}}_{\mathrm{s}}^{-},{\mathrm{N}}_{\mathrm{s}}^{+};{\mathrm{V}}^{-},{\mathrm{V}}^{\mathrm{o}} \)) are possible in principle, the foremost contribution to paramagnetism comes from only \( {\mathrm{N}}_{\mathrm{s}}^{\mathrm{o}} \). Let us therefore estimate the maximal possible concentration of NV in a DND on the basis of assumptions about the known total amounts of substitutional nitrogen and monovacancies inside the particles. The actual charge state of NV is not essential for such an estimation. A statistical approach gives the following simple formula for the probability (p NV ) of finding at least one NV in one 5-nm DND particle consisting of N nodes of covalent diamond lattice:\( {p}_{\mathrm{NV}}=\frac{2 nv}{N} \) , where n и v are the mean numbers of isolated nitrogen atoms and monovacancies inside the DND particle, respectively. Because N ПК ≈ 1300 ppm, we can approximately assume that n + v ~ 15 for N = 11,500. The maximal value of p NV is achieved when \( n\approx v\approx \frac{15}{2} \). This gives p NV ≈ 0.0098. This value corresponds to around ~ 1 ppm of NV in DND, as was obtained previously from comparison of the g = 4.27 EPR signals of the DND and the reference sample. Excluding the surface nodes linked with surface functional groups and interior nodes occupied by A-centres (up to 2 at.%) from the formal integration procedure, using N = 9950–10,000 gives a slightly greater concentration of NV, up to 1.1 ppm. The estimated experimental concentration of NV − centres in the DNDs is in close agreement with the theoretical estimation made above, and about 1000 times smaller than the concentration of all S = 1/2 paramagnetic species in the system.
The concentration ratio for interior S = 1/2\( {\mathrm{N}}_{\mathrm{s}}^{\mathrm{o}} \) and S = 1 NV − centres is therefore qualitatively consistent with the main idea of rapid NV centre formation, that is, from the “random inclusion” of both substitutional nitrogen atoms and vacancies to the growing diamond nano-crystallites during the overall ~ 13–20 μs duration of the detonation wave propagation. Here, we intuitively assume that each NV − is formed as a result of the random embedding and occurrence of impurity nitrogen atoms and vacancies in the nearest neighbour lattice sites during the period of time prior to the subsequent rapid cooling of the products to temperatures of the order of ~ 500–650 °C at which the diffusion of vacancies in the lattice is practically stopped.
Nitrogen Impurity Concentrations
XPS is a powerful tool for studying the DNDs’ composition and the chemical state of the main alien elements present on the surface of the DNDs (and also within 2 nm under the surface) [29]. Our main interest was focused on the XPS signal from nitrogen and the evaluation of the interior nitrogen content, since nitrogen is the predominant inner impurity. The XPS signals of carbon (C1s), nitrogen (N1s) and other elements have been recorded after etching of the surface with Ar ions. An overview of the XPS spectrum plotted in the wide range of binding energies from 250 to 600 eV is shown in Fig. 5a. Although the data indicates the presence of a large amount of oxygen-containing atomic groups at the DND’s surface (5.5 at.%), the analysis of the O1s signal is not of particular relevance to this work. The concentration of both inner and exterior nitrogen was preliminarily evaluated to be between 1.7 and 2.4 at.% [30]. The concentration of all the residual elements found (a small number of metals) did not exceed ~ 0.32 at.% in the as-received DND, and it could be reduced by 20–30 times by etching the DND powder in boiling aqua regia and hydrochloric acid [30]. The photoemission peak of nitrogen (N1s) is shown in Fig. 5b. Etching with Ar ions results in the removal of weakly bound adsorbed species from the surface, while the covalent diamond lattice remains unaffected. The characteristic high-energy peaks (~ 404.6 eV and 407.3 eV) in the N1s photoemission signal have very low intensities when compared with an untreated pristine sample. These peaks demonstrate the presence of any remaining nitrate ions (peak at ~ 407.3 eV) and nitrite groups (peak at ~ 404.6 eV) on the Ar ion-treated surface. Further complete removal of nitrate ions and nitrite groups from the surface can be achieved only by annealing the DND powder in air at temperature> 350 o C. The main peak of the N1s signal at ~ 401 eV, which is not influenced by Ar ion treatment, corresponds to chemical bonds of the type N–sp 3 –C. This peak is a characteristic of interior elemental nitrogen covalently bound within the diamond lattice. It appears due to various forms of nitrogen present in the diamond matrix, including NN dimers, next nearest neighbouring N + ...N − pairs, and more complex nitrogen clusters. Similar data were obtained for the photoemission peak of carbon C1s (sharp, intense signal seen in Fig. 5a, see also Ref. [30] for details). The C1s XPS signal consists of two main peaks:one centred at 284.9 eV corresponding to C–C bonds in the diamond matrix and another peak centred at 287.3 eV corresponding to C–N bonds. Only diamond carbon and sp 3 /sp 2 carbon bound to nitrogen, even in the form of atomic-scale disordered nitride phase species (where neighbouring carbon and nitrogen atoms can have up to three C–N bonds), are represented in the C1s signal of the Ar ion-treated surface. Our careful analysis of the integrated intensity of only the N1s 400.9 eV peak and C1s signals (together with O1s and residual elements’ signals) suggests that nitrogen is contained within the diamond lattice of selected DND sample in the amount of 1.65 ± 0.05 at.% and mainly in the form of complex clusters which are not paramagnetic. This value was obtained after special correction by a reducing factor taking into account that the actual size of DND particles is larger than the whole depth of crystal lattice from which the excited photoelectrons are readily emitted. It seems that only a small part of nitrogen is present in the diamond lattice in the form of isolated paramagnetic nitrogen atoms with spin S = 1/2 (no more than 8–9% of all nitrogen). The isolated paramagnetic nitrogen atoms are only accessible for observation by EPR as was shown in the previous section.

XPS spectra of acid-purified DNDs after Ar ion etching:a overview of the spectrum in the range 250-600 eV and b N1s photoemission peaks
XPS spectroscopy was also applied to estimate the nitrogen content in other DND samples provided by our suppliers. We found that the nitrogen content varies in these samples from ~ 1.6 at.% (minimal value) to ~ 2.1 at.% (maximal value). We simultaneously noticed that higher concentrations of nitrogen correspond to the samples with a smaller X-ray CSR size. Such values of nitrogen content were also roughly confirmed by elemental analysis with a Micro Corder JMC10 device in the course of sample combustion in oxygen flow (30 ml/min) at 1000 o C. As a reference source of nitrogen, carbon and hydrogen for this method, we used the antipyrine C11 H 12 N 2 O, having nitrogen in its structure in the form of N–N groups.
DND Fluorescence Intensity and Its Dependence upon the Nitrogen Content
The PL spectrum of one selected powder DND sample is shown in Fig. 6a. It has the maximum intensity at 680 nm. Following [31], a characteristic spectrum with peak position above 650 nm indicates the presence of both NV − and NV 0 defects, although the contribution from some light-emitting defective sites of remaining sp 2 -coordinated carbon species around DND particles cannot be excluded completely. However, the characteristic zero-phonon lines (ZPL) at 638 nm (NV − ) and 575 nm (NV 0 ), respectively, are not detected. This probably occurs due to the changing positions of the NV 0 and NV − excited/ground states inside the bandgap for centres lying in the vicinity of the particle edge and the subsequent broadening of the 638-nm and 575-nm ZPL spectral components for the ensembles of such NV 0 and NV − centres with slightly different electronic parameters. Let us mark that the absence of featured NV − ZPL peak at 638 nm was confirmed in many studies related with photoluminescence of DND aggregates lying free on the substrate or embedded inside polymers [31]. Sometimes it (suppressed or very poorly recognized ZPL feature at 638 nm) even happens for larger isolated fluorescent Ib HPHT nanodiamonds of a size about 40 nm [4].

а Photoluminescence spectrum of DND powder pressed flush in a shallow hole with a diameter of 2 mm made in a copper plate and b the interior nitrogen content measured by XPS and c the intensity of NV − PL at 680 nm as a function of X-ray CSR size for a series of selected DNDs synthesized in the presence of some intentionally added inorganic additives in the detonation zone (charge and water shell) as provided by the manufacturer. Excitation laser wavelength λ = 532 nm, power ~ 0.5 mW. The diameter of the focused laser spot on the sample surface was 2 μm. Conditions of recording were temperature T = 293 K, and an air environment
We also studied the PL intensity for a series of DND samples with different CSR sizes. The CSR size characterizes the average size of perfect diamond domains or the mean size of diamond nano-crystallites in the powder even if they are randomly arranged in large-scale polycrystalline aggregates with size exceeding 30–50 nm. The CSR size varied from 4.3 to 5.6 nm in the series of DND samples selected for our studies (similar to the results explained in Ref. [32] although we used another more traditional approach for analysing X-ray diffractograms). Figure 6b shows the dependence of interior nitrogen content evaluated by means of the XPS method versus the CSR size of the DNDs. The larger the CSR size, the smaller the nitrogen content. This seems reasonable as powders with larger DND particles were synthesized as a result of the addition of some inorganic substances having the elements playing the role of nitrogen-getter inside the detonation zone (charge or charge enclosure). Such elementary additives probably promote the reduction of the overall nitrogen content in the growing diamond lattice during the explosion process or change the conditions of the diamond lattice assembly to slightly prolong the synthesis (on the order of microseconds). In addition to the overall nitrogen content, we also recorded the reduced amount of nitrogen-related paramagnetic centres in these DNDs, as confirmed by EPR spectroscopy. PL spectra of all DNDs having different CSR sizes are practically the same in shape in the range 550–900 nm, but this is not the case for the absolute intensities of the PL at the maxima of the PL spectra at 650–680 nm. The intensity at the maximum of PL spectrum is plotted in Fig. 6c as a function of the X-ray CSR size of the DNDs. Comparing both panels (Fig. 6b, c) it is clearly seen that the smaller the nitrogen content in DND, the higher the NV − PL intensity. Again, this seems reasonable as nitrogen-related centres and especially A-centres can act as effective quenchers of PL if the NV − light-emitting centres in some DND particles (one per a hundred of 5 nm DND particles at least) are surrounded by a “gas” of A-centres and other lattice imperfections, similar to the approach proposed in Ref. [33]. This trend gives us a hint at possible ways to enhance the intensity of luminescence from ensembles of DND particles by manipulating their size and nitrogen concentration. Among them, there is the technological enlargement of the mean size of DND particles in the course of their treatment at high pressure and high temperature at appropriate conditions, promoting their recrystallization and further crystal growth [34]. The probable reduction of NV − content through treatment leading to the reduction of A-centres and other interior defects in the diamond lattice may be compensated in principle by the opposite trend, promoting the brightness of NV − emission, and reducing the amount of all types of PL quenchers in the system, and hence, substantially improving the crystal quality of particles with sizes exceeding a dozen nanometres [34, 35]. Further works on these topics are now in progress.
DND Aggregate-Specific Fluorescence and Discrimination of Diffraction-Limited Emitters
Characterizing the fluorescence from isolated DND particles or submicron aggregates is crucial both for understanding their potential use as fluorescent markers and to help to mitigate disadvantages related with a relatively low concentration of NV − in them as determined by EPR. Photoluminescence was recorded for DND aggregates spin-coated on a glass microscope coverslip from an aqueous suspension with an average size of DND aggregates about 30 nm (as measured by dynamic light scattering).
Figure 7a shows two PL 2D maps obtained by confocal microscopy, using a 532-nm wavelength excitation laser with 100-μW output optical power. Bright spots corresponding to DND aggregates up to 500–700 nm in lateral size are observed. Dimmer spots of size around the optical diffraction limit are also observed after selection of the appropriate isolated spots. Figure 7c shows the intensity distribution along the specially selected straight line aa plotted in Fig. 7a. This line crosses about six dim spots of smallest diameter—five spots (1–5) are crossed by the straight line fairly precisely along their centres and one spot (6) is with a small displacement from this line. The corresponding five peaks in intensity distribution are clearly seen in Fig. 7c. Thus, each dim spot 1–5 laying on the line aa corresponds to the DND aggregate of smallest lateral size (in the range below 70 nm). It is possible that all of them are fixed on one V-shaped straight groove existing on the glass coverslip. We successfully fitted each peak in the intensity distribution with a 2D Gaussian \( {A}_i{e}^{-\left[{\left(x-{x}_{oi}\right)}^2+{\left(y-{y}_{oi}\right)}^2\right]/2{s}_i^2} \), where x oi , y oi are the Cartesian coordinates of the centres of the dim round spots, A я -maximum PL intensity of each isolated spot, s я is a parameter close to s о ≈ r о /3, where s о is a 1/3 part of the Airy disk diameter r о . In our case, for λ = 532 nm radiation (in vacuum) and numerical aperture of microscopic objective NA = 1.40, we have the following values for r о и s о :r о =1.22λ/2NA ≈ 232 nm and s о ≈ 77 nm. The s о value in the 2D Gaussian determines the point spread function (PSF) of and ideal point-like emitter, i.e. the diffraction limitation related with the smallest possible interference ring. For the five peaks mentioned above (i = 1–5) we found the following s я values, respectively:85, 77, 77, 84 and 77 nm. Peak 5 has both the highest intensity and s -value equal to the theoretical value s о ≈ 77 nm. This means that the lateral size of the corresponding emitter for peak 5 is significantly smaller than the PSF size. The same is also true for peaks 1–4. We can conclude that the lateral sizes of DND aggregates laying along line aa are in the range below 70 nm. Each spherically shaped DND aggregate of ~ 30 nm in size Footnote 5 consisting of individual 5-nm particles (percentage of voids is 50%) has about 1.3 NV − centres in accordance with the 1.1 ppm content of NV − determined previously. This value is great enough in principle to distinguish aggregates of this size by optical methods. Each of the five aggregates lying along the line aa probably has between 2 and 10 colour centres. The larger the height of the DND aggregate lying on the substrate, the higher the density of luminescence centres (per unit square) for the light emitted more or less perpendicular to the surface. For aggregates with height up to 300–350 nm, the brightness of PL intensity can be at least one order of magnitude greater than that for aggregates of smaller 25–30 nm size. The PL spectrum of one selected aggregate of high brightness (marked by a circle in the 2D map presented in Fig. 7a) was studied in detail. It has roughly the same shape (especially on the right wing above 680 nm) as that for the spectrum shown in Fig. 6a and indicates the presence of both NV − and NV 0 дефекты. However, the characteristic zero-phonon lines (ZPL) at 638 nm (NV − ) and 575 nm (NV 0 ), respectively, were again not detected. ZPLs are usually well resolved only for diamond crystals of micron size or larger, while for nanometre-scale crystals, they are typically not well-observed due to some experimental or other physical reasons Footnote 6 . Let us mark that for DND aggregates lying on the cover glass, the final treatment with UV/O-Cleaner removed the sp 2 -coordinated carbon species around the particles inside the aggregates and as a result the PL spectrum is free from the contribution of light-emitting centres of disorganized sp 2 -carbon phase.

а 2D-colour mapping of the PL signal intensity of DND spin-coated on a glass microscope coverslip together with b schematics of the experimental setup. c The PL intensity profile along the “aa” cross-section. г The photoluminescence intensity versus time for the selected DND aggregate marked with a circle in the upper side of the 2D map shown in panel a , in the presence or absence of an external magnetic field. Laser excitation at 532 nm wavelength. Square, 201 × 201 pixels. Integration time is 3 ms/pixel. Step—100 nm. The excitation radiation is focused on the upper surface of the glass coverslip with deposited DND. In zero field (B = 0), only small changes of intensity, due to the blinking of some of the NV − colour centres occur. When magnetic field is temporally applied a large decrease of the PL intensity occurs. Upper left panel a :the left scale for PL intensity corresponds to the 2D mapping of another DND aggregate shown in the left bottom corner of the large 2D map. Upper right panel b :schematics of the experimental setup explaining the displacement of the permanent magnet above the coverslip relative to the deposited DND
To confirm the presence of NV − centres in the DND, we studied the influence of an external magnetic field on the PL intensity. No PL modification is expected from NV 0 , which have no magneto-optical properties unlike NV − . Figure 7d shows the meander-like time variation of the PL intensity from an isolated DND aggregate in the presence of an external magnetic field switched “ON” by quickly bringing a compact permanent magnet close to the DND or by removing it (“OFF”). This occurs as a result of the mixing of the |0〉GS and |−1〉GS states of NV − centres at the ground-state spin level anti-crossing (GSLAC), marked with a circle at magnetic field ~ 100 mT in Fig. 3. Such mixing leads to a change in the populations of these states, accompanied by a PL intensity decrease. The optical transitions between the ground 3 A2 and excited 2 E triplet states preserve the spin quantum number (Δm S = 0). However, from the m S = ± 1 excited state, the optical excitation of NV – also decays with no radiation in the visible domain, through a system of two metastable singlet states before coming back to the ground state [5]. This process is accompanied by radiation at a longer wavelength of 1042 nm defined by the gap between these two singlet levels with S = 0. This additional decay pathway results in a lower fluorescence intensity from the main radiation transition within the m S = ± 1 subsystem of ground and excited triplet states. The experimentally detected decrease of NV – centre PL intensity is quite reasonable in the presence of a weak (≤ 100 mT) magnetic field [36], as observed in Fig. 7d. Surprisingly, in our case (for DNDs), this drop is essentially larger than those reported in the literature for Ib HPHT fluorescent microdiamonds and even evaluated theoretically in the framework of the standard NV model [37].
Conclusion
In this article, we have shown that nitrogen-vacancy centres appeared in the interior of 5 nm nanodiamond particles synthesized by detonation of nitrogen-containing explosives are the main triplet colour centres. Precise counting of the number of NV − centres was achieved. The DNDs contain ~ 1.1 ppm of such NV − centres in the crystalline diamond lattice. This value is just five times smaller than that detected in the bright fluorescent reference Ib HPHT diamonds with a mean size of about ~ 100 nm. Our adjusted and rechecked estimation made on the basis of the EPR method is about three orders of magnitude larger than that evaluated in Ref. [38] by pure optical methods for DND aggregates. Probably not all NV − centres detected by EPR are optically active due to the NV − luminescence quenching by various point and collective defects located near the surface. Annealing DNDs in oxygen- or ozone-containing air removes defects associated with light-absorbing sp 2 carbon at the particle surface and allows detection of the specific PL of NV − centres. When an external magnetic field is applied, an accompanying variation of the PL intensity is observed. This is the result of the sensitivity of the NV − triplet ground state to magnetic field and comes through the mixing of the |0〉GS and |−1〉GS states of only the NV − centres below/near the GSLAC point leading to the decay of the optical excitation with no radiation in the visible domain. The application of this effect to discriminate ultra-small DND emitters in an environment with a large autofluorescence background and micron-scale bio-object contouring is promising [39]. Some DND aggregates playing the role of point-like NV − emitters and giving the smallest fluorescent spots with diameter close to the PSF size (diffraction limitation) were found.
Notes
- 1.
The same data for concentration of paramagnetic centres S = ½ (≥ 1260 ppm) were obtained independently by precision measurements of the magnetization field dependence at temperature T = 2 K using a superconducting SQUID magnetometer. The magnetization curve does not show the presence of spins 3/2 in appreciable quantity in DNDs [13].
- 2.
See for example the details of this method for analysis of the standard (DND) and non-standard (NDB-G) samples given in Ref. [22].
- 3.
Fluorescent Ib HPHT (high pressure high temperature) microcrystals doped with nitrogen (100–150 ppm) and thereafter irradiated by high energy electrons (2–15 MeV) and annealed at 800–950 o С.
- 4.
We assume that a typical DND particle (with size slightly exceeding 5 nm) consists of ~ 1.1 × 10 4 carbon atoms.
- 5.
Here we consider the typical DND aggregate in water DND suspension used for deposition the aggregates on the glass coverslip. Certainly, the actual shape of aggregates lying on the cover slip can be rougher and extremely diverse depending upon the adhesion of DND particles to the glass and actual size of the large-scale aggregates.
- 6.
The ZPL line at 638 nm was not resolved in many works where the nanodiamonds having NV − were studied [4, 31]. It is probable that the various local environments of NV − centres in nanosized diamond (like surface defects and interior imperfections) greatly disturb the position of NV − triplet ground states in the bandgap.
Сокращения
- 2D:
-
Two dimensional
- CCD:
-
Charge-coupled device
- DND:
-
Detonation nanodiamond
- EPR:
-
Electron paramagnetic resonance
- FND:
-
Fluorescent nanodiamond
- GSLAC:
-
Ground state spin levels anti-crossing
- HPHT:
-
High pressure high temperature
- NA:
-
Числовая апертура
- NV:
-
Nitrogen-vacancy
- ODMR:
-
Optically detectable magnetic resonance
- PL:
-
Фотолюминесценция
- PSF:
-
Point spread function
- SCTB:
-
Special design and technology bureau
- SQUID:
-
Superconducting quantum interference device
- UV:
-
Ультрафиолет
- XPS:
-
Рентгеновская фотоэлектронная спектроскопия
- XRD:
-
Рентгеновская дифракция
- ZPL:
-
Zero-phonon line
Наноматериалы
- Электрические поля и емкость
- Электромагнетизм
- Магнитные единицы измерения
- Магнитные поля и индуктивность
- Цифровые (ВКЛ / ВЫКЛ) устройства на эффекте Холла:переключатели и защелки
- Наноалмазы для магнитных датчиков
- Получение и магнитные свойства легированных кобальтом наночастиц шпинели FeMn2O4
- Модуляция свойств электронной и оптической анизотропии ML-GaS вертикальным электрическим полем
- (La0.97RE0.01Yb0.02) 2O2S Нанофосфор, преобразованный из слоистого гидроксилсульфата, и исследование фотолюминесценции…
- Что такое магнитное экранирование?