


Моделирование и имитация предлагает новые идеи для SARS-CoV-2
В нашем предыдущем блоге мы обсуждали использование инструментов прогнозного моделирования для построения начальных структур на атомном уровне потенциальных мишеней для лекарств (например, белков) и для уточнения областей, которые не поддаются экспериментальному определению ( см. Видео ). Эти инструменты включают добавление атомов водорода и гибких петель, которые иногда невозможно разрешить экспериментально. Мы исследовали это в контексте криоэлектронной микроскопии (крио-ЭМ) белка шипа (S) SARS-CoV-2, недавно опубликованного в журнале Science (DOI:10.1126 / science.abb2507).
В этом блоге мы подробно расскажем, как молекулярное моделирование и симуляция уточненных структурных моделей, таких как белок SARS-CoV-2 S, может помочь в генерации новых гипотез для открытия и разработки предполагаемых терапевтических средств для лечения COVID-19.
Связывание лекарств зависит от структурных изменений
В живых системах белки естественным образом существуют как динамические сущности. Их динамика часто предопределяет их функцию. Физик Ричард Фейнман однажды сказал:
«Если бы мы назвали самое сильное предположение из всех, которое ведет к бесконечным попыткам понять жизнь, это то, что все вещи состоят из атомов и что все, что делают живые существа, можно понять в терминах покачивания и покачивания атомов. " 1
Белок SARS-CoV-2 S не является исключением из принципа Фейнмана. Перед тем как попасть в клетки человека, белок S связывает рецептор, называемый ангиотензинпревращающим ферментом 2 (ACE2). 2 Рецептор-связывающий домен (RBD) является частью белка S, который связывает ACE2 ( Рисунок 1A ). RBD может существовать по крайней мере в двух первичных конформационных состояниях, называемых верхним (рецептор доступен) и нижним (рецептор недоступен) состояниями ( Рисунок 1B ). Когда RBD находится в активном состоянии, белок S более «открыт» для облегчения связывания ACE2. 2 Исследования показали, что нижнее состояние, недоступное для рецепторов, более стабильно. 2 Это означает, что предполагаемые терапевтические средства, такие как небольшие органические молекулы, способные связывать RBD, могут стабилизировать RBD в неактивном состоянии и предотвращать взаимодействие вируса с ACE2; таким образом, предотвращая заражение людей COVID-19.
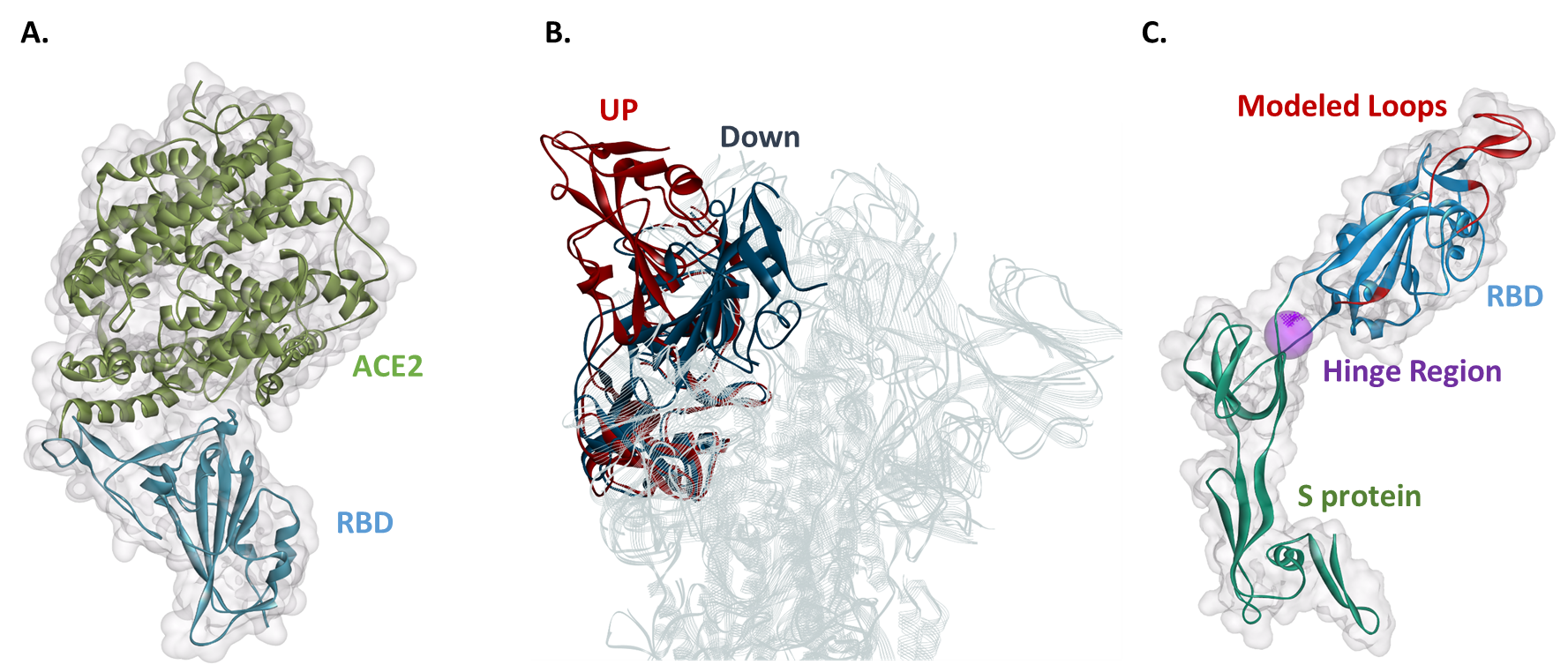
СРП S-белка похожа на петлю на двери
Гибкий линкер соединяет RBD с оставшимся белком S. Гибкость компоновщика позволяет RBD переходить из нижнего состояния в верхнее посредством движения сгибания шарнира ( Рис. 1B ). Из белка SARS-CoV-2 S мы извлекли RBD с соответствующим гибким линкером и соседним доменом ( Рисунок 1C ). Мы использовали структуру RBD из белка S в активном состоянии (PDB 6VYB), но в этой структуре отсутствуют три петли, потенциально важные для связывания ACE2 ( Рисунок 1C ). В результате нам пришлось построить модель гомологии RBD с гибким линкером, используя крио-ЭМ структуру, из которой было выполнено усечение (PDB 6VYB) с дополнительным шаблоном. Дополнительным шаблоном была кристаллическая структура только RBD в комплексе с ACE2 (PDB 6M17) ( Рисунок 1A ).
Структура RBD содержит петли, включающие один более чем 20 аминокислотных остатков, которых нет в структуре открытого состояния (PDB 6VYB). Две из этих петель образуют взаимодействие с ACE2; таким образом, моделирование гомологии необходимо для понимания взаимодействий белка Spike. Затем мы можем назначить атомы водорода в соответствующих состояниях протонирования, имитирующих физиологические условия, такие как pH.
Затем мы могли бы выполнить моделирование молекулярной динамики (МД), чтобы смоделировать конформационный переход и / или предсказать возможные сайты связывания, где предполагаемые небольшие молекулы могут связываться, чтобы нарушить взаимодействие S-белка с ACE2. Когда мы предсказали возможные сайты связывания с помощью BIOVIA Discovery Studio для нашей модели гомологии RBD с гибким линкером, мы идентифицировали сайт связывания, расположенный на шарнире гибкого линкера ( Рисунок 1C ). Мы назвали эту область шарниром и отметили, что она заслуживает дальнейшего исследования для открытия предполагаемых терапевтических средств. Если бы небольшая молекула связалась в шарнирной области, она, вероятно, могла бы заблокировать RBD в неактивном состоянии и, таким образом, предотвратить связывание ACE2.
Дальнейшие расследования
Обширные исследования, включая компьютерные предсказания и биологические эксперименты, могут еще больше прояснить полезность шарнирной области. Примеры вычислительных прогнозов могут включать анализ в нормальном режиме (NMA) и / или моделирование MD. 3 Например, длительный временной масштаб, примерно сотни наносекунд, моделирования МД может позволить ученым выбрать несколько конформаций шарнирной области.
С другой стороны, NMA может обеспечить грубую и быструю оценку конформационных переходов. 3 Конкретные конформации из моделирования MD и / или NMA являются отправными точками для высокопроизводительного виртуального скрининга потенциальных баз данных малых молекул. Ученые могли состыковывать и оценивать каждую маленькую молекулу по всем конформациям. Затем они могли оценить все полученные позы и представить лучшие результаты для экспериментальной проверки. Исследования показали, что этот метод компьютерного открытия лекарств, часто называемый виртуальным скринингом на основе ансамблей, увеличивает шансы на выявление кандидатов на лекарства. 4 Этот метод также отражает тот факт, что связывание лекарств зависит от структурных изменений белка, как отмечалось выше. Мы считаем, что приведенные здесь предварительные результаты интересны и заслуживают дальнейшего изучения.
Во-вторых, мы хотели бы отметить, что уточненная структурная модель S-белка может быть использована в качестве мишени для иммунотерапевтов. 5 Ученые могли разработать моноклональные антитела, которые связываются с белками SARS CoV-2 S, основываясь на предварительных знаниях о сайте связывания ACE2. Затем они могли выполнять in silico исследования созревания аффинности для улучшения специфичности связывания. 6
Как активный сторонник научного сообщества, которое сегодня сотрудничает в области решений COVID-19, BIOVIA Dassault Systèmes разрабатывает BIOVIA Discovery Studio. Эта проверенная среда моделирования и симуляции наук о жизни объединяет более 30 лет рецензируемых исследований и первоклассные in silico техники. Программное обеспечение предоставляет ученым полный набор инструментов для использования от идентификации цели до оптимизации потенциальных клиентов, включая инструменты для дизайна и анализа биологических препаратов, классического моделирования, дизайна на основе структуры и фрагмента, виртуального скрининга лигандов, а также ADME и прогнозирования токсичности. P>
В рамках корпоративной социальной ответственности Dassault Systèmes BIOVIA рада предложить квалификационным академическим исследовательским группам, участвующим в исследованиях, связанных с SARS-CoV-2, бесплатную шестимесячную лицензию на BIOVIA Discovery Studio чтобы помочь им в поиске быстрых, безопасных и эффективных терапевтических препаратов-кандидатов против вируса SARS-CoV-2. Если вы академический исследователь в этой области, запросите лицензию на программное обеспечение и загрузите ее. Это предложение действует до 30 июня 2020 г.
Биопрепараты
- Pixus:новые толстые и прочные лицевые панели для встроенных плат
- GE представляет новый продукт для приложений управления и мониторинга
- DSM и Nedcam для разработки новых приложений для 3D-печати большого размера
- Teradyne планирует новый Cobot Hub для портфельных компаний UR и MiR
- PLASTICS представляет новый стандарт безопасности для робототехники и литья под давлением
- Новая дорожная карта для цепочек поставок нефти и газа
- Коучинг для устойчивого развития:внедрение и поддержка новых процессов и изменений
- B&R представляет новый инструмент моделирования для разработки цифровых двойников
- АББ обеспечивает планирование автоматизации и электрификации для нового рудника в Швеции
- 5G и Edge поднимают новые задачи кибербезопасности на 2021 год